Berg J.M., Tymoczko J.L., Stryer L. Biochemistry
Подождите немного. Документ загружается.

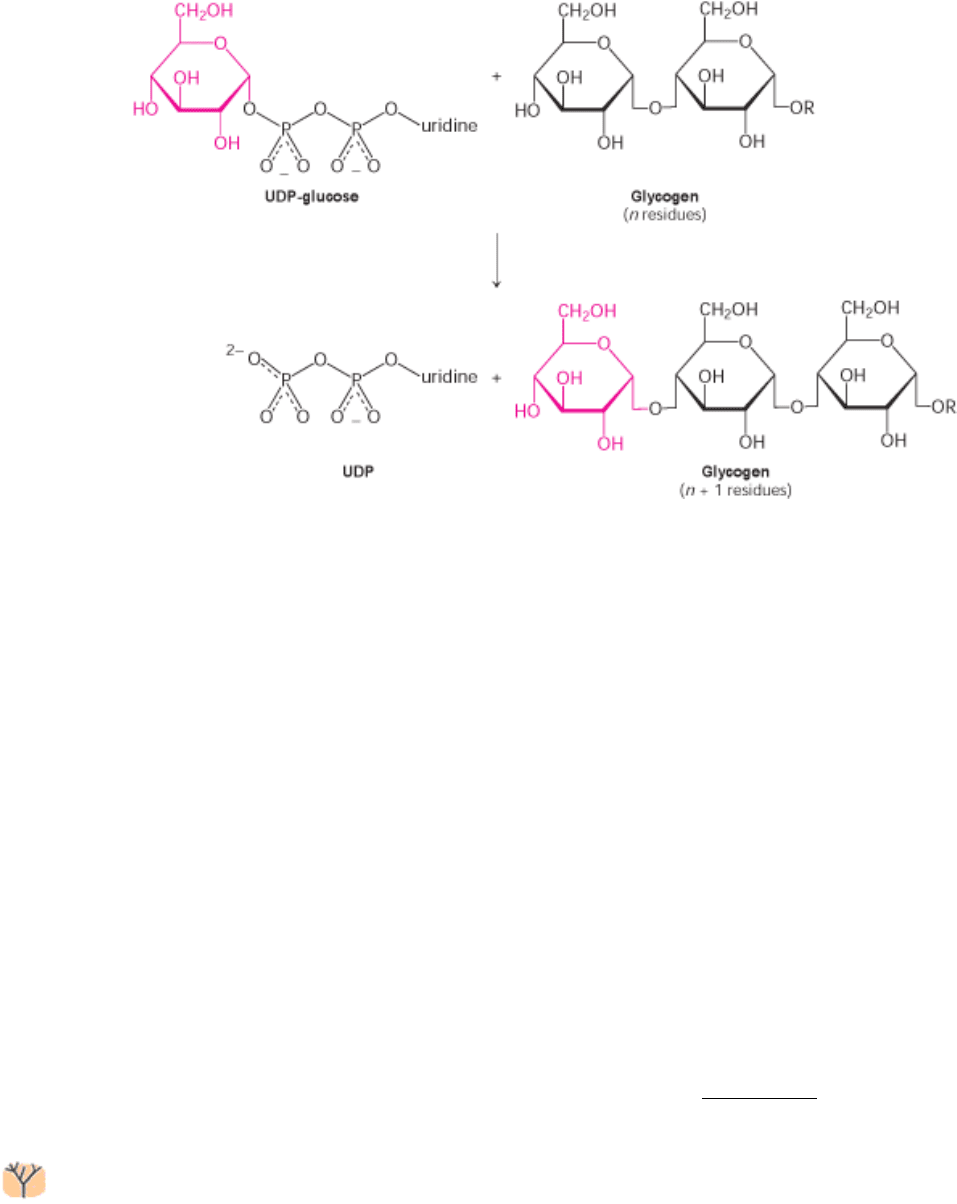
21.4.2. Glycogen Synthase Catalyzes the Transfer of Glucose from UDP-Glucose to a
Growing Chain
New glucosyl units are added to the nonreducing terminal residues of glycogen. The activated glucosyl unit of UDP-
glucose is transferred to the hydroxyl group at a C-4 terminus of glycogen to form an α-1,4-glycosidic linkage. In
elongation, UDP is displaced by the terminal hydroxyl group of the growing glycogen molecule. This reaction is
catalyzed by glycogen synthase, the key regulatory enzyme in glycogen synthesis.
Glycogen synthase can add glucosyl residues only if the polysaccharide chain already contains more than four residues.
Thus, glycogen synthesis requires a primer. This priming function is carried out by glycogenin, a protein composed of
two identical 37-kd subunits, each bearing an oligosaccharide of α-1,4-glucose units. Carbon 1 of the first unit of this
chain, the reducing end, is covalently attached to the phenolic hydroxyl group of a specific tyrosine in each glycogenin
subunit. How is this chain formed? Each subunit of glycogenin catalyzes the addition of eight glucose units to its partner
in the glycogenin dimer. UDP-glucose is the donor in this autoglycosylation. At this point, glycogen synthase takes over
to extend the glycogen molecule.
21.4.3. A Branching Enzyme Forms α-1,6 Linkages
Glycogen synthase catalyzes only the synthesis of α-1,4 linkages. Another enzyme is required to form the α-1,6 linkages
that make glycogen a branched polymer. Branching occurs after a number of glucosyl residues are joined in α-1,4
linkage by glycogen synthase. A branch is created by the breaking of an α-1,4 link and the formation of an α-1,6 link:
this reaction is different from debranching. A block of residues, typically 7 in number, is transferred to a more interior
site. The branching enzyme that catalyzes this reaction is quite exacting. The block of 7 or so residues must include the
nonreducing terminus and come from a chain at least 11 residues long. In addition, the new branch point must be at least
4 residues away from a preexisting one.
Branching is important because it increases the solubility of glycogen. Furthermore, branching creates a large number of
terminal residues, the sites of action of glycogen phosphorylase and synthase (Figure 21.15). Thus, branching increases
the rate of glycogen synthesis and degradation.
Glycogen branching requires a single transferase activity. Glycogen debranching requires two enzyme activities: a
transferase and an α-1,6 glucosidase. Sequence analysis suggests that the two transferases and, perhaps, the α-1,6

glucosidase are members of the same enzyme family, termed the α -amylase family. Such an enzyme catalyzes a reaction
by forming a covalent intermediate attached to a conserved aspartate residue (Figure 21.16). Thus, the branching enzyme
appears to function through the transfer of a chain of glucose molecules from an α-1,4 linkage to an aspartate residue on
the enzyme and then from this site to a more interior location on the glycogen molecule to form an α-1,6 linkage.
21.4.4. Glycogen Synthase Is the Key Regulatory Enzyme in Glycogen Synthesis
The activity of glycogen synthase, like that of phosphorylase, is regulated by covalent modification. Glycogen synthase
is phosphorylated at multiple sites by protein kinase A and several other kinases. The resulting alteration of the charges
in the protein lead to its inactivation (Figure 21.17). Phosphorylation has opposite effects on the enzymatic activities of
glycogen synthase and phosphorylase. Phosphorylation converts the active a form of the synthase into a usually inactive
b form. The phosphorylated b form requires a high level of the allosteric activator glucose 6-phosphate for activity,
whereas the a form is active whether or not glucose 6-phosphate is present.
21.4.5. Glycogen Is an Efficient Storage Form of Glucose
What is the cost of converting glucose 6-phosphate into glycogen and back into glucose 6-phosphate? The pertinent
reactions have already been described, except for reaction 5, which is the regeneration of UTP. ATP phosphorylates
UDP in a reaction catalyzed by nucleoside diphosphokinase.
Thus, one ATP is hydrolyed incorporating glucose 6-phosphate into glycogen. The energy yield from the breakdown of
glycogen is highly efficient. About 90% of the residues are phosphorolytically cleaved to glucose 1-phosphate, which is
converted at no cost into glucose 6-phosphate. The other 10% are branch residues, which are hydrolytically cleaved. One
molecule of ATP is then used to phosphorylate each of these glucose molecules to glucose 6-phosphate. The complete
oxidation of glucose 6-phosphate yields about 31 molecules of ATP, and storage consumes slightly more than one
molecule of ATP per molecule of glucose 6-phosphate; so the overall efficiency of storage is nearly 97%.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.4. Glycogen Is Synthesized and Degraded by Different Pathways
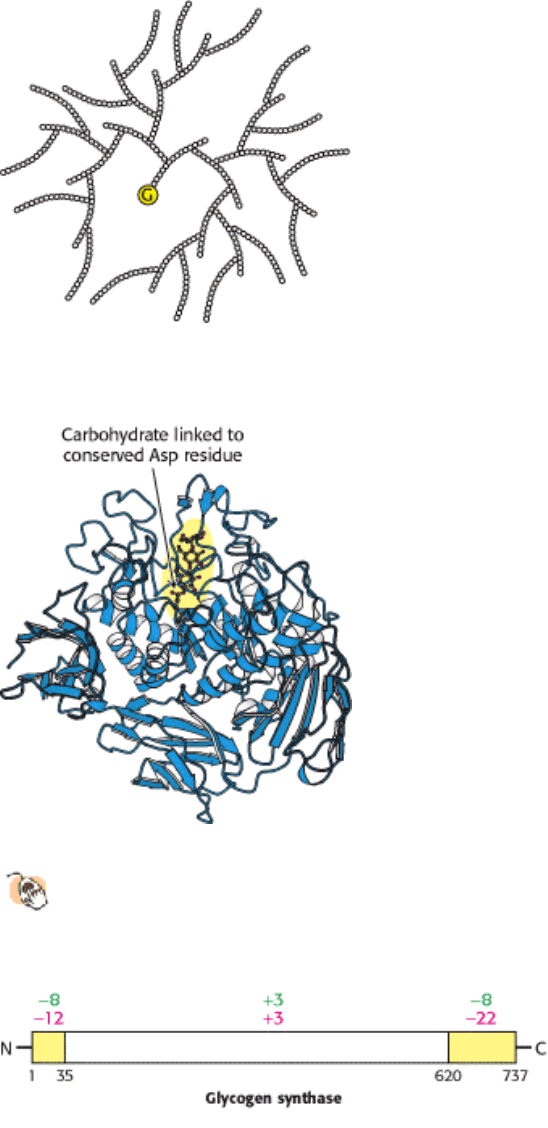
Figure 21.15. Cross Section of a Glycogen Molecule. The component labeled G is glycogenin.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.4. Glycogen Is Synthesized and Degraded by Different Pathways
Figure 21.16. Structure of Glycogen Transferase.
A conserved aspartate residue forms a covalent intermediate with a
chain of glucose molecules.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.4. Glycogen Is Synthesized and Degraded by Different Pathways
Figure 21.17. Charge Distribution of Glycogen Synthase. Glycogen synthase has a highly asymmetric charge
distribution. Phosphorylation markedly changes the net charge of the amino- and carboxyl-terminal regions (yellow) of
the enzyme. The net charge of these regions and the interior of the enzyme before and after complete phosphorylation
are shown in green and red, respectively. [After M. F. Browner, K. Nakano, A. G. Bang, and R. J. Fletterick. Proc. Natl.
Acad. Sci. USA 86(1989):1443.]

II. Transducing and Storing Energy 21. Glycogen Metabolism
21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
We now return to the regulation of glycogen metabolism with a knowledge of both degradation and synthesis. Glycogen
breakdown and synthesis are reciprocally regulated by a hormone-triggered cAMP cascade acting through protein
kinase A (Figure 21.18). In addition to phosphorylating and activating phosphorylase kinase, protein kinase A adds a
phosphoryl group to glycogen synthase, which leads to a decrease in enzymatic activity. This important control
mechanism prevents glycogen from being synthesized at the same time that it is being broken down. How is the
enzymatic activity reversed so that glycogen breakdown halts and glycogen synthesis begins?
21.5.1. Protein Phosphatase 1 Reverses the Regulatory Effects of Kinases on Glycogen
Metabolism
The changes in enzymatic activity produced by protein kinases are reversed by protein phosphatases. The hydrolysis of
phosphorylated serine and threonine residues in proteins is catalyzed by protein phosphatases. One enzyme, termed
protein phosphatase 1, plays key roles in regulating glycogen metabolism. PP1 inactivates phosphorylase kinase and
phosphorylase a by dephosphorylating these enzymes. PP1 decreases the rate of glycogen breakdown: it reverses the
effects of the phosphorylation cascade. Moreover, PP1 also removes the phosphoryl group from glycogen synthase b to
convert it into the much more active a form. Hence, PP1 accelerates glycogen synthesis. PP1 is yet another molecular
device for coordinating carbohydrate storage.
The complete complex of PP1 consists of three components: PP1 itself, a 37-kd catalytic subunit; a 123-kd R
Gl
subunit
that confers high affinity for glycogen; and inhibitor 1, a small regulatory subunit that, when phosphorylated, inhibits
PP1. The importance of the R
Gl
subunit is that it brings PP1, which is active only when associated with glycogen
molecules, into proximity with its substrates.
How is the phosphatase activity of PP1 itself regulated? Consider the case in which glycogen degradation is predominant
(Figure 21.19). In this case, PKA is active. Two components of PP1 are themselves substrates for protein kinase A.
Phosphorylation of the R
Gl
component by protein kinase A prevents R
Gl
from binding the catalytic subunit of PP1.
Consequently, activation of the cAMP cascade leads to the inactivation of PP1 because it can no longer bind its
substrates. Phosphorylation of inhibitor 1 by protein kinase A blocks catalysis by PP1. Thus, when glycogen degradation
is switched on by cAMP, the accompanying phosphorylation of inhibitor 1 keeps phosphorylase in the active a form and
glycogen synthase in the inactive b form. The epinephrine-induced phosphorylation of the R
Gl
subunit and inhibitor 1 are
complementary devices for sustaining glycogen degradation.
21.5.2. Insulin Stimulates Glycogen Synthesis by Activating Protein Phosphatase 1
How is glycogen synthesis stimulated? As stated earlier, the presence of glucagon signifies the starved state and initiates
glycogen breakdown while inhibiting glycogen synthesis. When blood-glucose levels are high, insulin stimulates the
synthesis of glycogen by triggering a pathway that activates protein phosphatase 1 (Figure 21.20). The first step in the
action of insulin is its binding to a receptor tyrosine kinase in the plasma membrane. Multiple phosphorylations again
serve as the instigation for a regulatory wave of dephosphorylations. The binding of insulin to its receptor leads to the
activation of an insulin-sensitive protein kinase that phosphorylates the R
Gl
subunit of PP1 at a site different from that
modified by protein kinase A. This phosphorylation leads to the association of the R
Gl
subunit with PP1 and the
glycogen molecule. The consequent dephosphorylation of glycogen synthase, phosphorylase kinase, and phosphorylase
promotes glycogen synthesis and blocks its degradation. Once again we see that glycogen synthesis and breakdown are
coordinately controlled.
21.5.3. Glycogen Metabolism in the Liver Regulates the Blood-Glucose Level

After a meal rich in carbohydrates, blood-glucose levels rise, leading to an increase in glycogen synthesis in the liver.
Although insulin is the primary signal for glycogen synthesis, other, nonhormonal mechanisms also function in the liver.
One signal is the concentration of glucose in the blood, which normally ranges from about 80 to 120 mg per 100 ml (4.4
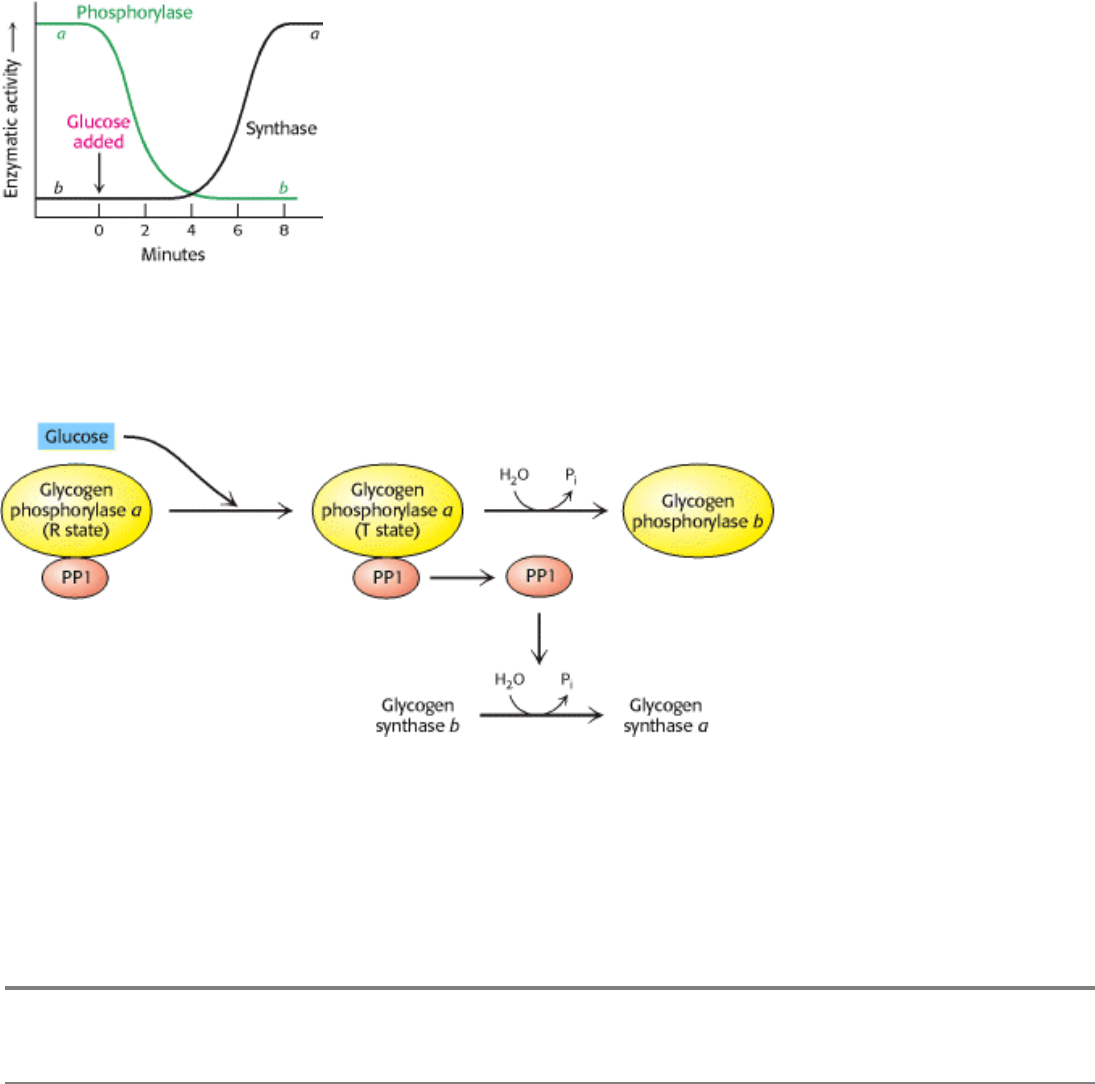
6.7 mM). The liver senses the concentration of glucose in the blood and takes up or releases glucose accordingly. The
amount of liver phosphorylase a decreases rapidly when glucose is infused (Figure 21.21). After a lag period, the amount
of glycogen synthase a increases, which results in the synthesis of glycogen. In fact, phosphorylase a is the glucose
sensor in liver cells. The binding of glucose to phosphorylase a shifts its allosteric equilibrium from the active R form to
the inactive T form. This conformational change renders the phosphoryl group on serine 14 a substrate for protein
phosphatase 1. It is significant that PP1 binds tightly to phosphorylase a but acts catalytically only when glucose induces
the transition to the T form. Recall that the R T transition of muscle phosphorylase a is unaffected by glucose and is
thus unaffected by the rise in blood-glucose levels (Section 21.2.2).
How does glucose activate glycogen synthase? Phosphorylase b, in contrast with phosphorylase a, does not bind the
phosphatase. Consequently, the conversion of a into b is accompanied by the release of PP1, which is then free to
activate glycogen synthase (Figure 21.22). Removal of the phosphoryl group of inactive glycogen synthase b converts it
into the active a form. Initially, there are about 10 phosphorylase a molecules per molecule of phosphatase. Hence, the
activity of glycogen synthase begins to increase only after most of phosphorylase a is converted into b. This remarkable
glucose-sensing system depends on three key elements: (1) communication between the serine phosphate and the
allosteric site for glucose, (2) the use of PP1 to inactivate phosphorylase and activate glycogen synthase, and (3) the
binding of the phosphatase to phosphorylase a to prevent the premature activation of glycogen synthase.
21.5.4. A Biochemical Understanding of Glycogen-Storage Diseases Is Possible
Edgar von Gierke described the first glycogen-storage disease in 1929. A patient with this disease has a huge
abdomen caused by a massive enlargement of the liver. There is a pronounced hypoglycemia between meals.
Furthermore, the blood-glucose level does not rise on administration of epinephrine and glucagon. An infant with this
glycogen-storage disease may have convulsions because of the low blood-glucose level.
The enzymatic defect in von Gierke disease was elucidated in 1952 by Carl and Gerty Cori. They found that glucose 6-
phosphatase is missing from the liver of a patient with this disease. This was the first demonstration of an inherited
deficiency of a liver enzyme. The liver glycogen is normal in structure but present in abnormally large amounts. The
absence of glucose 6-phosphatase in the liver causes hypoglycemia because glucose cannot be formed from glucose 6-
phosphate. This phosphorylated sugar does not leave the liver, because it cannot cross the plasma membrane. The
presence of excess glucose 6-phosphate triggers an increase in glycolysis in the liver, leading to a high level of lactate
and pyruvate in the blood. Patients who have von Gierke disease also have an increased dependence on fat metabolism.
This disease can also be produced by a mutation in the gene that encodes the glucose 6-phosphate transporter. Recall
that glucose 6-phosphate must be transported into the lumen of the endoplasmic reticulum to be hydrolyzed by
phosphatase (Section 16.3.5). Mutations in the other three essential proteins of this system can likewise lead to von
Gierke disease.
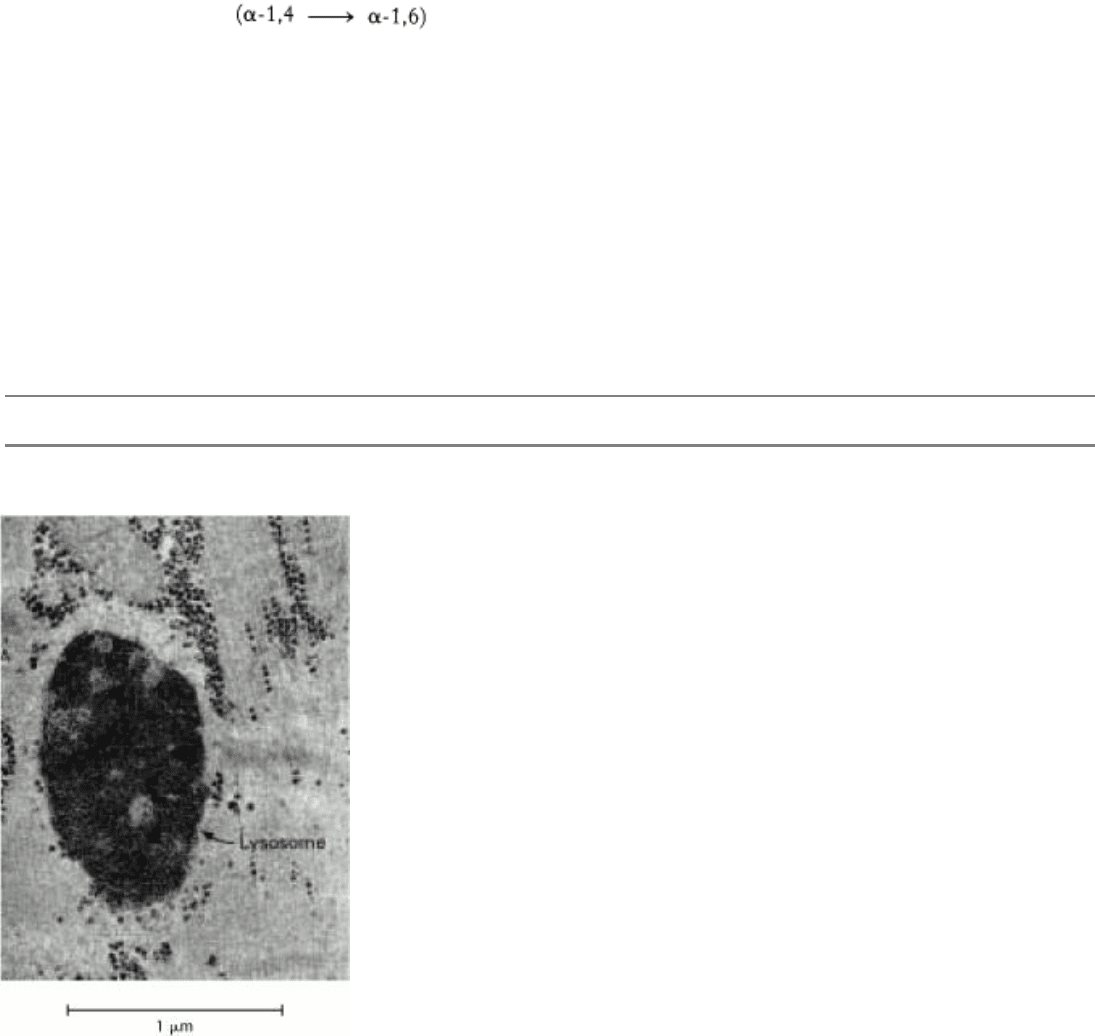
Seven other glycogen-storage diseases have been characterized (Table 21.1). In Pompe disease (type II), lysosomes
become engorged with glycogen because they lack α-1,4-glucosidase, a hydrolytic enzyme confined to these organelles
(Figure 21.23). The Coris elucidated the biochemical defect in another glycogen-storage disease (type III), which cannot
be distinguished from von Gierke disease (type I) by physical examination alone. In type III disease, the structure of liver
and muscle glycogen is abnormal and the amount is markedly increased. Most striking, the outer branches of the
glycogen are very short. Patients having this type lack the debranching enzyme (α-1,6-glucosidase), and so only the
outermost branches of glycogen can be effectively utilized. Thus, only a small fraction of this abnormal glycogen is
functionally active as an accessible store of glucose.
A defect in glycogen metabolism confined to muscle is found in McArdle disease (type V). Muscle phosphorylase
activity is absent, and the patient's capacity to perform strenuous exercise is limited because of painful muscle cramps.
The patient is otherwise normal and well developed. Thus, effective utilization of muscle glycogen is not essential for
life. The results of phosphorus-31 nuclear magnetic resonance studies of these patients have been very informative. The
pH of skeletal muscle cells of normal people drops during strenuous exercise because of the production of lactate. In
contrast, the muscle cells of patients with McArdle disease become more alkaline during exercise because of the
breakdown of creatine phosphate (Section 14.1.5). Lactate does not accumulate in these patients because the glycolytic
rate of their muscle is much lower than normal; their glycogen cannot be mobilized. The results of NMR studies have
also shown that the painful cramps in this disease are correlated with high levels of ADP (Figure 21.24). NMR
spectroscopy is a valuable, noninvasive technique for assessing dietary and exercise therapy for this disease.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
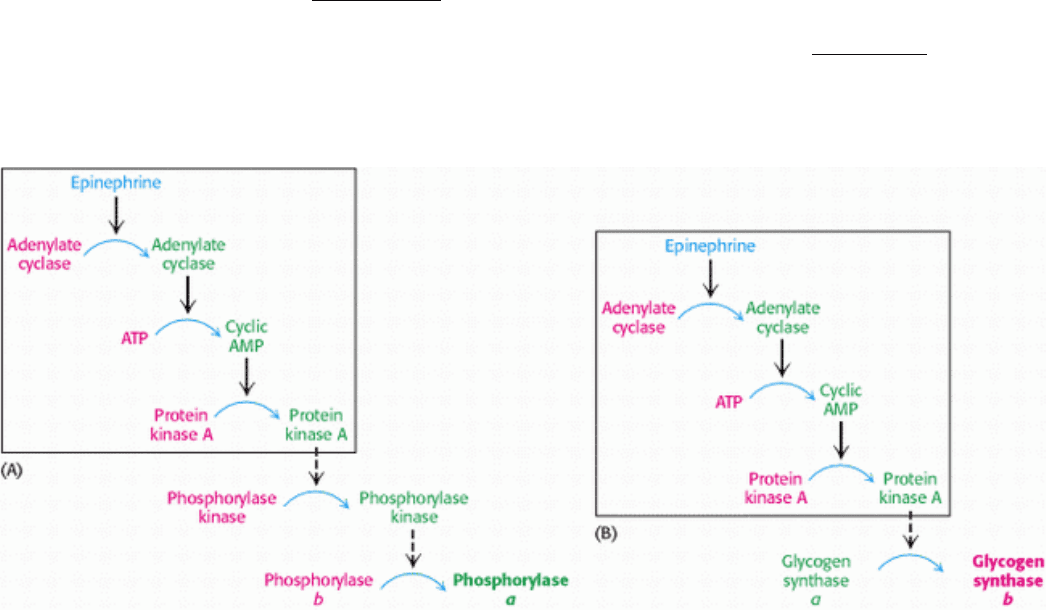
Figure 21.18. Coordinate Control of Glycogen Metabolism. Glycogen metabolism is regulated, in part, by hormone-
triggered cyclic AMP cascades: (A) glycogen degradation; (B) glycogen synthesis. Inactive forms are shown in red, and
active ones in green. The sequence of reactions leading to the activation of protein kinase A is the same in the regulation
of glycogen degradation and synthesis. Phosphorylase kinase also inactivates glycogen synthase.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
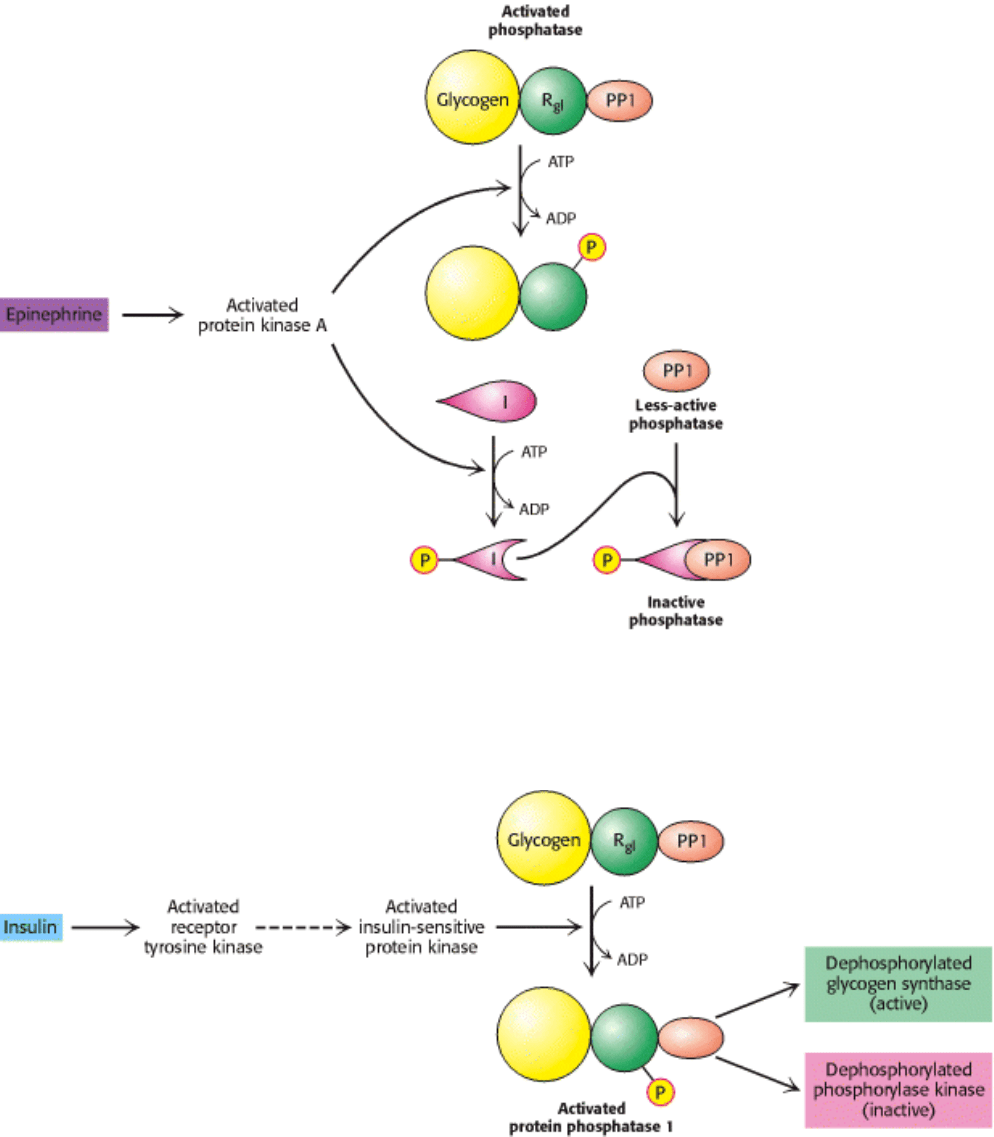
Figure 21.19. Regulation of Protein Phosphatase 1 (PP1). Phosphorylation of R
Gl
by protein kinase A dissociates the
catalytic subunit from the glycogen particle and hence the PP1 substrates. Inhibition is complete when the inhibitor
subunit (I) is phosphorylated and binds to PP1 to inactivate it.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Figure 21.20. Insulin Activates Protein Phosphatase 1. Insulin triggers a cascade leading to the activation of protein
phosphatase 1, which results in the stimulation of glycogen synthesis and inhibition of its breakdown. The activated
receptor tyrosine kinase switches on a putative master kinase that phosphorylates the insulin-sensitive protein kinase. In
turn, the glycogen-targeting subunit (R
Gl
subunit) of the phosphatase is phosphorylated, which activates the enzyme.
[After P. Dent, A. Lavoinne, S. Nakielny, F. B. Caudwell, P. Watt, and P. Cohen. Nature 348(1990):306.]
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Figure 21.21. Blood Glucose Regulates Liver Glycogen Metabolism. The infusion of glucose into the bloodstream
leads to the inactivation of phosphorylase, followed by the activation of glycogen synthase, in the liver. [After W.
Stalmans, H. De Wulf, L. Hue, and H.-G. Hers. Eur. J. Biochem. 41(1974):127.]
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Figure 21.22. Glucose Regulation of Liver Glycogen Metabolism. Glucose binds to and inhibits glycogen
phosphorylase a in the liver, leading to the dissociation and activation of protein phosphatase 1 (PP1) from glycogen
phosphorylase a. The free PP1 dephosphorylates glycogen phosphorylase a and glycogen synthase b, leading to the
inactivation of glycogen breakdown and the activation of glycogen synthesis.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Table 21.1. Glycogen-storage diseases
Type Defective enzyme Organ
affected
Glycogen in the
affected organ
Clinical features
I Von Gierke
disease
Glucose 6-phosphatase or transport
system
Liver and
kidney
Increased amount;
normal structure.
Massive enlargement of the
liver. Failure to thrive.
Severe hypoglycemia,
ketosis, hyperuricemia,
hyperlipemia.
II Pompe
disease
α-1,4-Glucosidase (lysosomal)
All organs Massive increase
in amount; normal
structure.
Cardiorespiratory failure
causes death, usually before
age 2.
III Cori disease Amylo-1,6-glucosidase
(debranching enzyme)
Muscle and
liver
Increased amount;
short outer
branches.
Like type I, but milder
course.
IV Andersen
disease
Branching enzyme
Liver and
spleen
Normal amount;
very long outer
branches.
Progressive cirrhosis of the
liver. Liver failure causes
death, usually before age 2.
V McArdle
disease
Phosphorylase Muscle Moderately
increased amount;
normal structure.
Limited ability to perform
strenuous exercise because of
painful muscle cramps.
Otherwise patient is normal
and well developed.
VI Hers disease Phosphorylase Liver Increased amount. Like type I, but milder
course.
VII Phosphofructokinase Muscle Increased amount;
normal structure.
Like type V.
VIII Phosphorylase kinase Liver Increased amount;
normal structure.
Mild liver enlargement. Mild
hypoglycemia.
Note: Types I through VII are inherited as autosomal recessives. Type VIII is sex linked.
II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Figure 21.23. Glycogen-Engorged Lysosome. This electron micrograph shows skeletal muscle from an infant with type
II glycogen-storage disease (Pompe disease). The lysosomes are filled with glycogen because of a deficiency in α-1,4-
glucosidase, a hydrolytic enzyme confined to lysosomes. The amount of glycogen in the cytosol is normal. [From H.-G.
Hers and F. Van Hoof, Eds. Lysosomes and Storage Diseases (Academic Press, 1973), p. 205.]

II. Transducing and Storing Energy 21. Glycogen Metabolism 21.5. Glycogen Breakdown and Synthesis Are Reciprocally Regulated
Figure 21.24. NMR Study of Human Arm Muscle. The level of ADP during exercise increases much more in a patient
with McArdle glycogen-storage disease (type V) than in normal controls. [After G. K. Radda. Biochem. Soc. Trans. 14
(1986):522.]
II. Transducing and Storing Energy 21. Glycogen Metabolism
Summary
Glycogen, a readily mobilized fuel store, is a branched polymer of glucose residues. Most of the glucose units in
glycogen are linked by α-1,4 glycosidic bonds. At about every tenth residue, a branch is created by an α-1,6-glycosidic
bond. Glycogen is present in large amounts in muscle cells and in liver cells, where it is stored in the cytoplasm in the
form of hydrated granules.
Glycogen Breakdown Requires the Interplay of Several Enzymes
Most of the glycogen molecule is degraded to glucose 1-phosphate by the action of glycogen phosphorylase, the key
enzyme in glycogen breakdown. The glycosidic linkage between C-1 of a terminal residue and C-4 of the adjacent one is
split by orthophosphate to give glucose 1-phosphate, which can be reversibly converted into glucose 6-phosphate.
Branch points are degraded by the concerted action of an oligosaccharide transferase and an α-1,6-glucosidase.
Phosphorylase Is Regulated by Allosteric Interactions and Reversible Phosphorylation
Phosphorylase is regulated by allosteric effectors and reversible covalent modifications. Phosphorylase b, which is
usually inactive, is converted into active phosphorylase a by the phosphorylation of a single serine residue in each
subunit. This reaction is catalyzed by phosphorylase kinase. The b form in muscle can also be activated by the binding of
AMP, an effect antagonized by ATP and glucose 6-phosphate. The a form in the liver is inhibited by glucose. The AMP-
binding sites and phosphorylation sites are located at the subunit interface. In muscle, phosphorylase is activated to
generate glucose for use inside the cell as a fuel for contractile activity. In contrast, liver phosphorylase is activated to
liberate glucose for export to other organs, such as skeletal muscle and the brain.
Epinephrine and Glucagon Signal the Need for Glycogen Breakdown
Epinephrine and glucagon stimulate glycogen breakdown through specific 7TM receptors. Muscle is the primary target
of epinephrine, whereas the liver is responsive to glucagon. Both signal molecules initiate a kinase cascade that leads to
the activation of glycogen phosphorylase.
Glycogen Is Synthesized and Degraded by Different Pathways