Ortiz de Montellano Paul R.(Ed.) Cytochrome P450. Structure, Mechanism, and Biochemistry
Подождите немного. Документ загружается.


Inhibition of Cytochrome P450 Enzymes
285
for these reactions but experimental evidence to
support the mechanisms is not available.
The P450 destruction mediated by halocarbons
was once believed to stem from the secondary
action of the lipid peroxides that are concomi-
tantly formed, but it is now evident that sub-
stances like CCI4 can destroy the heme group
directly^^^'
^^^' 423^26
J^IQ
associated cross-linking
of heme fragments to the protein suggests that a
radical species (CCI3 or CCI3O2) may be respon-
sible for heme destruction^^ ^ On the other hand,
the catalytic reduction of halocarbons, including
CCI4,
produces semistable complexes with
Soret maxima in the 450-500 nm range'^^^"^^^
Studies with model iron porphyrins, including the
detailed characterization of a dichlorocarbene-
metalloporphyrin complex"^^^, suggest that the
long-wavelength Soret bands are due to ferrous
halocarbene-heme complexes. This hypothesis is
supported by the finding that carbon monoxide is
formed in the reductive metabolism of CCI4 by
P450.
This reaction is likely to occur by a mecha-
nism similar to that for the generation of carbon
monoxide from methylenedioxyphenyl complexes
(Section 3.2.1)"^^^. In fact, porphyrin dichlorocar-
bene-iron complexes do react with water to give
carbon monoxide and with primary amines to give
isonitriles"^^^'
^^^.
Studies with halothane suggest
that it is also possible to form complexes in which
the halocarbon is a-bonded to ferric heme iron
atom435-^37
The links between formation of an iron-alkyl
complex and irreversible destruction of the heme
moiety have not been forged, but model studies
with diaryl- and carbethoxy-substituted carbene
complexes suggest that the halogenated carbenes
may shift to form a bond with a nitrogen of the
porphyrin'^^^^'^^ The resulting A/-haloalkyl adduct
are likely to undergo water-dependent hydrolysis
and might therefore not be detected by the methods
used to isolate other iV-alkyl porphyrins. However,
the formation of alternative reactive species that
attack the protein or the heme cannot be ruled out.
High (1-5 mM) concentrations of indo-
methacin and other nonsteroidal anti-inflamma-
tory agents reportedly denature P450 enzymes
because of their surfactant properties"^"^^. The
loss of P450 content seen when indomethacin is
added to liver microsomes is paralleled by essen-
tially stoichiometric appearance of a P420 peak.
Although it is likely that other agents cause P450
denaturation, it is unlikely that the process is phys-
iologically relevant because of the high drug
concentrations that are required.
4. P450 Enzyme Specificity
The isoform-specific inhibition of P450
enzymes is a promising avenue for the develop-
ment of therapeutic, insecticidal, and herbicidal
agents, as well as for investigation of the struc-
tures,
mechanisms, and biological roles of
individual P450 enzymes. The biosynthetic P450
enzymes have been the primary focus of efforts
to develop isoform-specific P450 inhibitors
because (a) they are better targets for specific
inhibitors because of their high substrate speci-
ficity and (b) there is high practical utility for such
inhibitors. In contrast, the broad, overlapping,
specificities of xenobiotic metabolizing P450
isoforms makes the design of isoform-specific
rather than -selective inhibitors more dififi-
cult'^'^^' ^^. Selective inhibitors of P450 enzymes,
as illustrated by the amphetamines^^^' ^^^,
TA0236,237,244-246^ Secobarbital^^' ^^\ gestodene^^^
ftirafylline^^^'
^^2,
1-ethynylpyrene^^' ^^, and
2,3',4,5'-tetramethoxystilbene'*'^^ are fairly com-
mon (see Appendix). Caution is required in
evaluating claims of inhibitor selectivity or speci-
ficity, as they are limited by the range of P450
enzymes actually examined. Only in the case where
an inhibitor has been tested with all the known
P450 isoforms in an organism can it be truly said to
be specific, at least in that organism. The claim for
specificity of inhibitors tested against only two or
three isoforms is necessarily limited.
5. Inhibitors of Biosynthetic
Enzymes
Several comprehensive review articles have
discussed the potential clinical relevance and
applications of inhibitors of biosynthetic P450
enzymes^' 446-448 jj^^ following discussion will
therefore be limited to an illustration of the
strategies employed in the design and develop-
ment of the currently available and/or prospective
inhibitors and their mechanistic diversity.
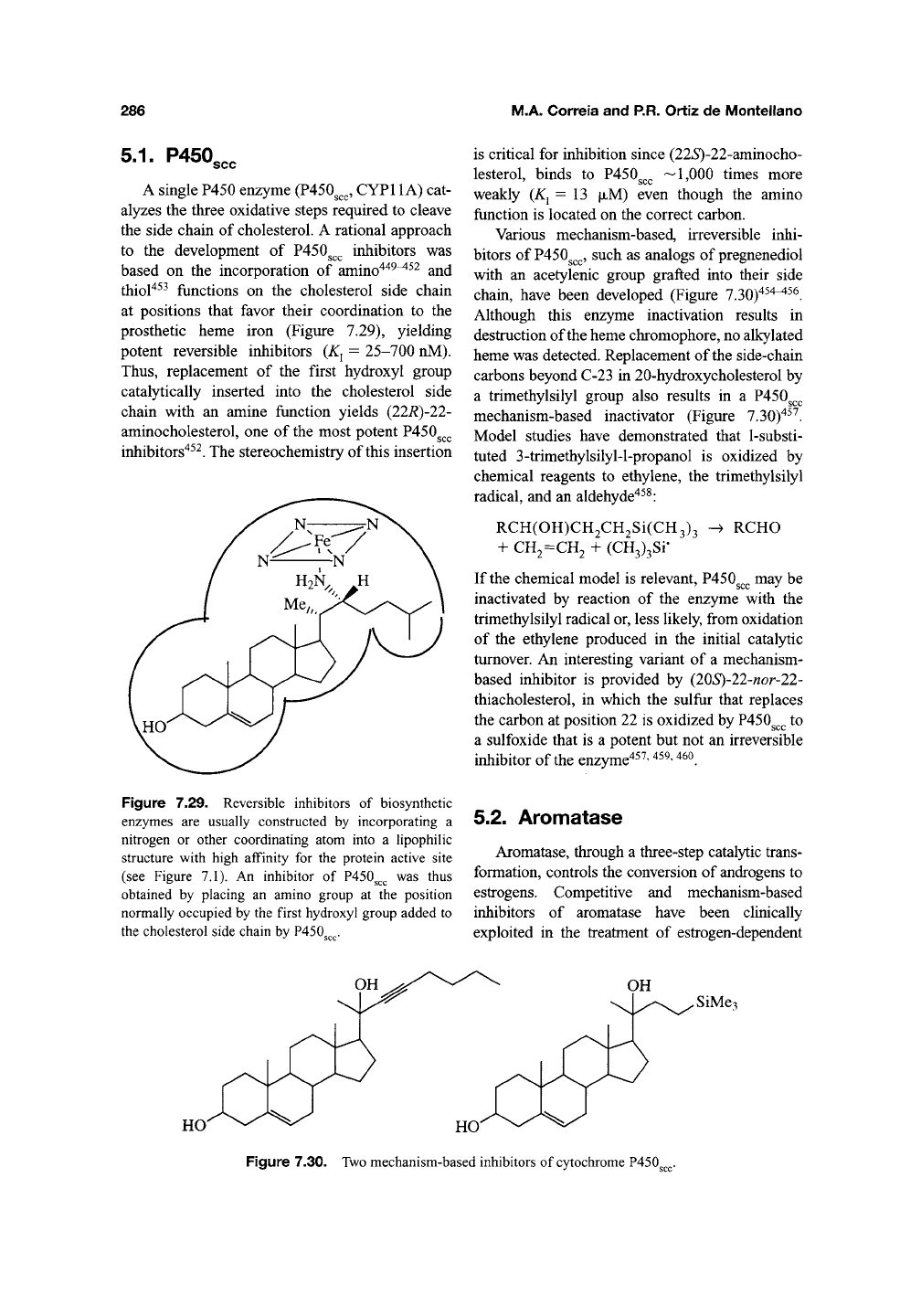
286
M.A.
Correia
and P.R.
Ortiz
de
Montellano
5.1.
P450,,,
A single P450 enzyme (P450^^^, CYPl 1 A) cat-
alyzes the three oxidative steps required to cleave
the side chain of cholesterol. A rational approach
to the development of P450g^^ inhibitors was
based on the incorporation of amino"^"^^^^^ and
thiol^^^ functions on the cholesterol side chain
at positions that favor their coordination to the
prosthetic heme iron (Figure 7.29), yielding
potent reversible inhibitors
(K^
= 25-700 nM).
Thus,
replacement of the first hydroxyl group
catalytically inserted into the cholesterol side
chain with an amine function yields (22R)-22-
aminocholesterol, one of the most potent P450g^^
inhibitors"^^^. The stereochemistry of this insertion
is critical for inhibition since (225)-22-aminocho-
lesterol, binds to P450g^^ —1,000 times more
weakly (^i = 13
|LJLM)
even though the amino
function is located on the correct carbon.
Various mechanism-based, irreversible inhi-
bitors of
P450g^^,
such as analogs of pregnenediol
with an acetylenic group grafted into their side
chain, have been developed (Figure 7.30)^^^^^^^.
Although this enzyme inactivation results in
destruction of the heme chromophore, no alkylated
heme was detected. Replacement of the side-chain
carbons beyond C-23 in 20-hydroxycholesterol by
a trimethylsilyl group also results in a P450g^^
mechanism-based inactivator (Figure 7.30)^^^.
Model studies have demonstrated that
1-substi-
tuted 3-trimethylsilyl-l-propanol is oxidized by
chemical reagents to ethylene, the trimethylsilyl
radical, and an aldehyde"^^^:
RCH(OH)CH2CH2Si(CH3)3 -> RCHO
+ CH2=CH2 + (CH3)3Si-
If the chemical model is relevant, P450g^^ may be
inactivated by reaction of the enzyme with the
trimethylsilyl radical or, less likely, from oxidation
of the ethylene produced in the initial catalytic
turnover. An interesting variant of a mechanism-
based inhibitor is provided by (20»S)-22-«or-22-
thiacholesterol, in which the sulfur that replaces
the carbon at position 22 is oxidized by P450g^^ to
a sulfoxide that is a potent but not an irreversible
inhibitor of the enzyme^^^' ^59,46o
Figure
7.29.
Reversible
inhibitors
of
biosynthetic
enzymes
are
usually
constructed
by
incorporating
a
nitrogen
or
other
coordinating
atom
into
a
hpophiHc
structure
with high
affinity
for the
protein
active
site
(see
Figure
7,1). An
inhibitor
of P450^^^ was
thus
obtained
by
placing
an
amino
group
at the
position
normally
occupied
by the
first
hydroxyl
group
added
to
the
cholesterol
side
chain
by
P450
5.2. Aromatase
Aromatase, through a three-step catalytic trans-
formation, controls the conversion of androgens to
estrogens. Competitive and mechanism-based
inhibitors of aromatase have been clinically
exploited in the treatment of estrogen-dependent
HO
SiMe^
Figure
7.30. Two
mechanism-based
inhibitors
of
cytochrome
P450
Inhibition of Cytochrome P450 Enzymes
287
mammary tumors^' ^61^66
^j^^
benign prostatic
hyperplasia"^^^'
^^^,
and have some promise in the
control of coronary heart disease"^^^. Indeed, some
of the more promising newer agents are in cUnical
^j^^jg463^66 Aminoglutethimide, an inhibitor of
aro-
matase, has been used to treat hormone-dependent
metastatic breast carcinoma, but its poor specificity
and the incidence of side effects, primarily
from inhibition of P450g^^, has compromised its
utility^^^^^^. Replacement of the aminophenyl
group in aminoglutethimide (Figure 7.31) by a
p)nidine moiety affords [pyridoaminoglutethimide
(3-ethyl-3-(4-pyridyl) piperidine-2,6-dione)], an
agent that inhibits aromatase but not P450g^^'^^^'
'*^^. The enhanced specificity for aromatase may
reflect a differential positioning of the pyridyl
nitrogen within the two active sites that enables it
to coordinate with the heme of aromatase but not
of P450g^^. Interestingly, an aminoglutethimide
analog with a nitrogen at the opposite end is a
more potent P450g^^ inhibitor than glutethimide
but has little or no activity against aromatase^^^.
These findings suggest that the 19-methyl and
carbon 22 of the sterol side chain are separated by
a distance roughly equal to the length of the
aminoglutethimide structure. It is therefore
tempting to speculate that aminoglutethimide
binds within the active sites of P450 and
sec
P450^j.Q^ in approximately the same orientation as
the normal sterol substrate, and that the location
of the nitrogen in the two inhibitors dictates their
differential enzyme selectivity.
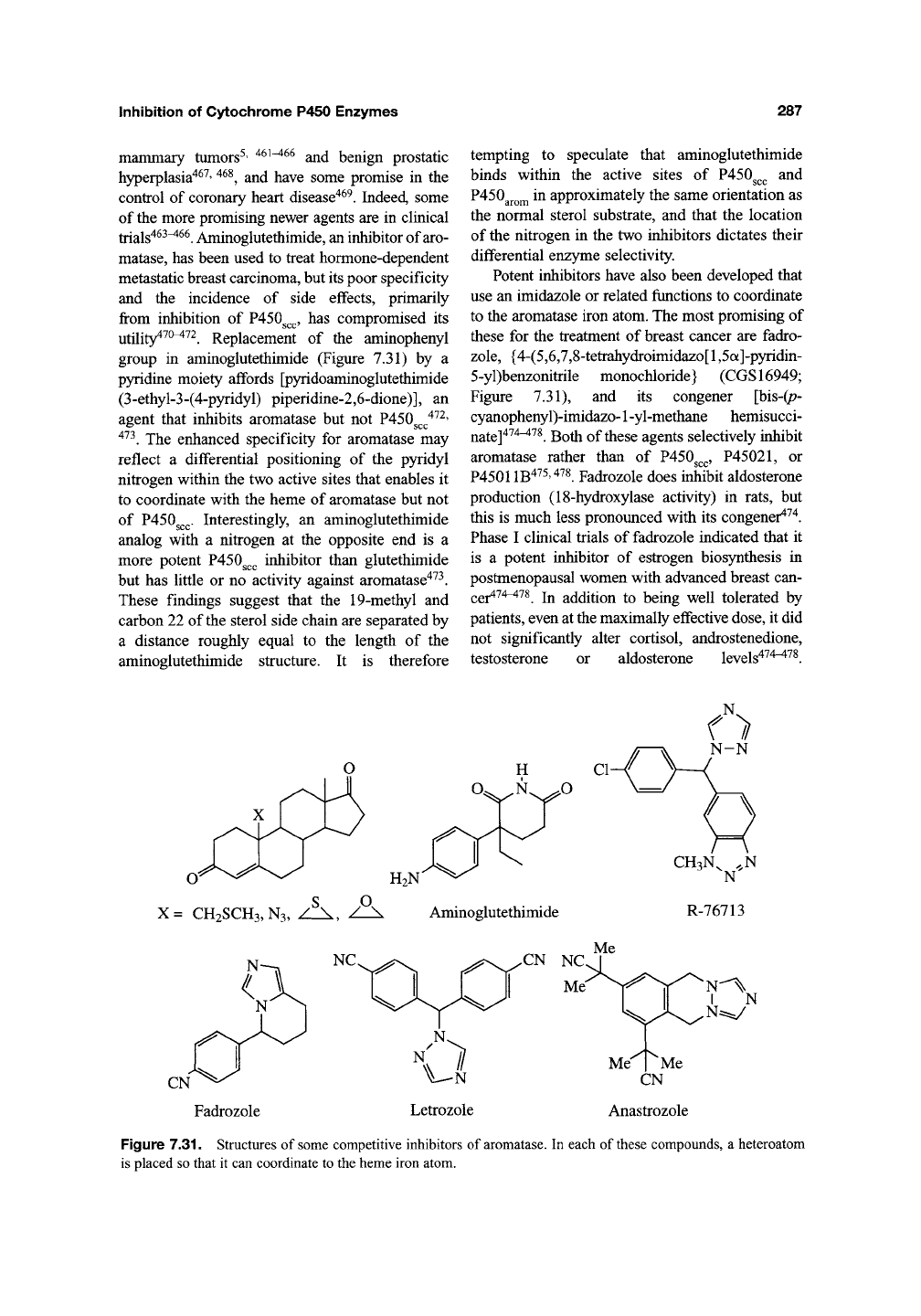
Potent inhibitors have also been developed that
use an imidazole or related functions to coordinate
to the aromatase iron atom. The most promising of
these for the treatment of breast cancer are fadro-
zole,
{4-(5,6,7,8-tetrahydroimidazo[l ,5a]-pyridin-
5-yl)benzonitrile monochloride} (CGS16949;
Figure 7.31), and its congener [bis-(p-
cyanophenyl)-imidazo-1 -yl-methane hemisucci-
nate]^^^^^^.
Both of these agents selectively inhibit
aromatase rather than of P450g^,^,
P45021,
or
P45011B475,478 Fadrozole does inhibit aldosterone
production (18-hydroxylase activity) in rats, but
this is much less pronounced with its congener^^^.
Phase I clinical trials of fadrozole indicated that it
is a potent inhibitor of estrogen biosynthesis in
postmenopausal women with advanced breast can-
^gj474-478 jj^ addition to being well tolerated by
patients, even at the maximally effective dose, it did
not significantly alter Cortisol, androstenedione,
testosterone or aldosterone levels"^^"^^^.
H2N
S O
X= CH2SCH3,N3, / \, / \
Aminoglutethimide
CH3N, .,N
N
R-76713
Fadrozole
:%:
N^
N
N^y
Me Me
CN
Anastrozole
Figure 7.31. Structures of some competitive inhibitors of aromatase. In each of these compounds, a heteroatom
is placed so that it can coordinate to the heme iron atom.
288
M.A. Correia and P.R. Ortiz de Montellano
However, fadrozole may now be surpassed by letro-
zole (CGS20267 or Femara; Figure 7.31), an
advanced nonsteroidal aromatase inhibitor, which
appears to be more potent and effective than fadro-
zole in the treatment of postmenopausal women
with advanced breast cancer^^^'
'*^^.
6 [(4-Chlorophenyl)( 1H-1,2,4-triazol-1 -yl)
methyl]-1 -methyl-1 H-benzotriazole, (R76713;
Figure 7.31), is a relatively selective and very
potent inhibitor of human placental aro-
matase"^^ ^~^^^. Its (+) enantiomer has a lower IC5Q
value than the (-) enantiomer when assayed
against human placental aromatase and exhibits
no appreciable inhibition of other steroidogenic
enzymes or liver microsomal P450 enzymes at
concentrations up to
1,000-times
the aromatase
Tp 482,483
Several other nonsteroidal compounds have
been developed as novel and selective aromatase
inhibitors, including 4-(4'-aminobenzyl)-2-oxazo-
lidinones"^^"^, 7-(alpha-azolylbenzyl)-1 H-indoles
and indolines of which l-ethyl-7-[(imidazol-l-yl)
(4-chlorophenyl)methyl]-lH-indole 12c exhibited
the most promising potency*^^, 4-imidazolylfla-
vans"^^^,
and anastrozole (Arimidex; Figure 7.31)"^^^.
Of these anastrozole has recently been approved
in the United States and several other countries for
the adjuvant treatment of postmenopausal women
with hormone receptor-positive early breast
cancer^^^'
^^^.
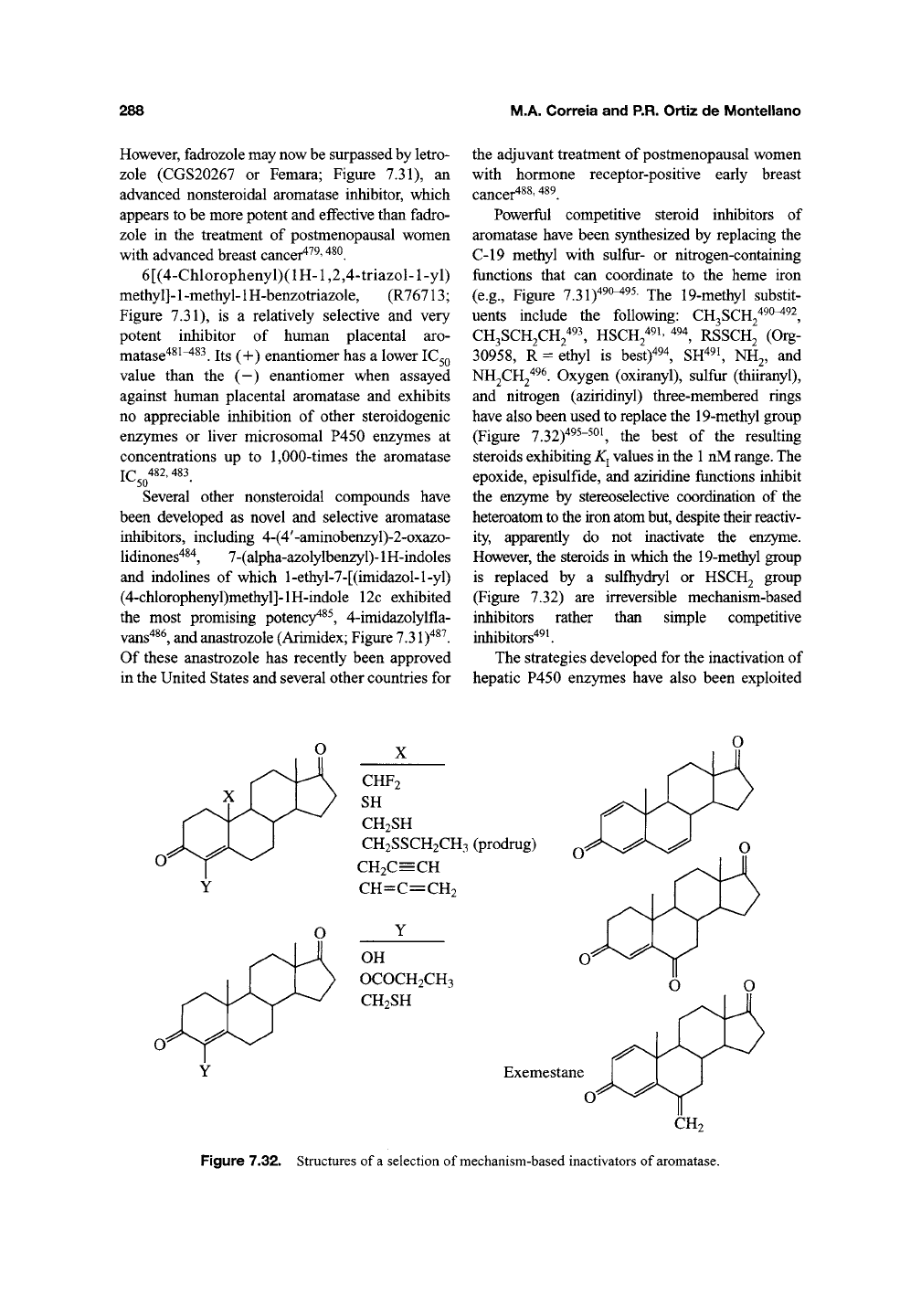
Powerful competitive steroid inhibitors of
aromatase have been synthesized by replacing the
C-19 methyl with sulfur- or nitrogen-containing
functions that can coordinate to the heme iron
(e.g.. Figure 7.31)^^^^^^ The 19-methyl substit-
uents include the following: CH^SCR^'^^^^^,
CH3SCH2CH/93^ HSCH2^9i, 494^ RSSCH2 (Org-
30958,
R = ethyl is best)^^^, SH^^i, NH2, and
NH2CH2^^^ Oxygen (oxiranyl), sulfur (thiiranyl),
and nitrogen (aziridinyl) three-membered rings
have also been used to replace the 19-methyl group
(Figure 7.32)^^^-^^^ the best of the resulting
steroids exhibiting
K^
values in the
1
nM
range.
The
epoxide, episulfide, and aziridine functions inhibit
the enzyme by stereoselective coordination of the
heteroatom to the iron atom but, despite their reactiv-
ity, apparently do not inactivate the enzyme.
However, the steroids in wiiich the 19-methyl group
is replaced by a sulfliydryl or HSCH2 group
(Figure 7.32) are irreversible mechanism-based
inhibitors rather than simple competitive
inhibitors'*^*.
The strategies developed for the inactivation of
hepatic P450 enzymes have also been exploited
CHF2
SH
CH2SH
CH2SSCH2CH3 (prodrug)
CH2C=CH
CH=C=CH2
OH
OCOCH2CH3
CH2SH
Exemestane
CH2
Figure 7.32. Structures of
a
selection of mechanism-based inactivators of
aromatase.

Inhibition of Cytochrome P450 Enzymes
289
in the design and synthesis of mechanism-based
aromatase inactivators. Substitution of the nor-
mally hydroxylated methyl group (C-19) with a
propargylic or allenic moiety (Figure 7.32) con-
verts the sterol into an irreversible aromatase inhi-
bitor^^^"^^^. The details of aromatase inactivation
by these acetylenic and allenic agents remain
unclear, but it is likely that they are activated
to intermediates that react with either the heme
or the protein (see Sections 3.1 and 3.3.2).
Replacement of the C-19 methyl with a difluo-
romethyl also yields a mechanism-based inactiva-
tor that must be activated by C-19 hydroxylation
(Figure 7.32)^^^^^^ as tritium release from the tri-
tium-labeled difluoromethyl derivative is required
for enzyme inactivation^^^. It is likely that the
difluoromethylalcohol thus produced decomposes
to the acyl fluoride that irreversibly binds to a pro-
tein nucleophile.
The 19-substituted analog of androst-4-ene-3,
17 dione steroid inhibitors, Org-30958 [19-
(ethyldithio)androst-4-ene-3,17-dione], has been
assessed in Phase I clinical trials for estrogen-
dependent breast cancer chemotherapy^^'*. The
ethyldithio substitution apparently renders the
steroid more stable extracellularly than the free
thiol Org-30365 (19-mercapto-androst-4-ene-3,
17-dione), resulting in vivo in animal models in an
8-fold
greater aromatase inhibitory activity than
either 4-OHA or SH-489. Its in vivo potency
requires intracellular reduction of the disulfide to
release the 19-mercapto analog Org-30365, a
more potent mechanism-based human placental
aromatase inactivator"^^^ than 4-OHA or
SH489494.
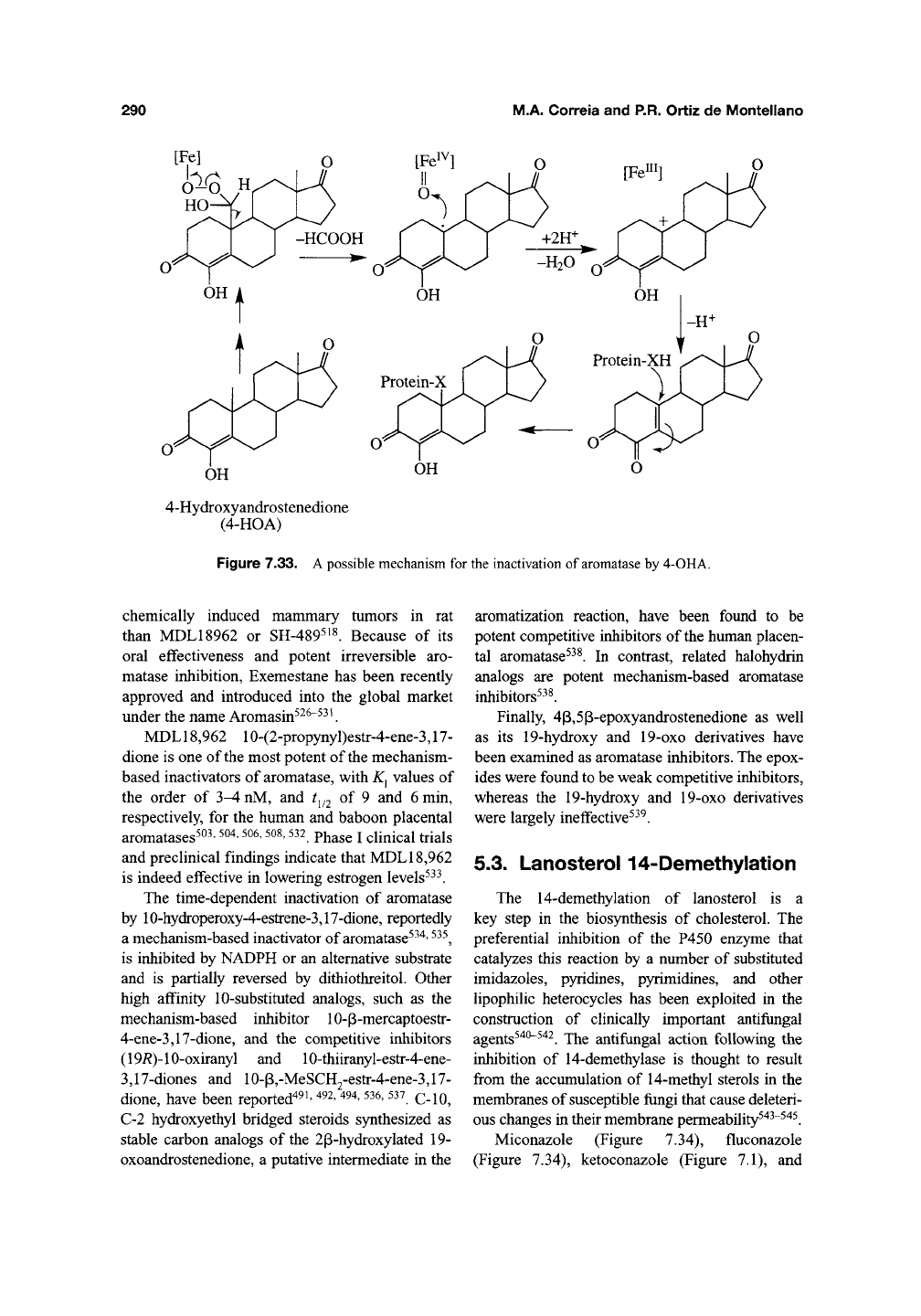
Clii^ically effective mechanism-based aro-
matase inactivators can also be obtained by intro-
ducing substituents at the 4- or 6-positions of the
sterol skeleton. 4-Acetoxy- and 4-hydroxy-
4-androstene-3,17-dione (4-OHA) (Figure 7.32)
irreversibly inactivate placental aromatase by
catalysis-dependent mechanisms involving the 19-
methyl^^^'
^^'*.
A possible mechanism for inhibition
of aromatase by the 4-substituted analogs, as illus-
trated by 4-OHA, is shown in Figure 7.33. 4-OHA
is used for the treatment of estrogen-dependent
breast cancer^^^' ^^^. Of a series of A^'^, A"^'^,
and A^'^ analogs evaluated as prospective aro-
matase inhibitors in preclinical trials, FC 24928
(4-aminoandrostan-l,4,6-triene-3,l7-dione) is the
most promising candidate because it inactivates
human placental aromatase activity as potently as
4-OHA and FCE-24304 (6-methylene-androstan-
l,4,-diene-3,l7-dione) but, unlike both these
compounds, it has little intrinsic androgenic
activity and does not affect 5a-reductase or
P450 516-518^
sec
Conjugation of the 4-hydroxyandrostene
nucleus as in 1,4,6 androstatriene-3,17-dione
(ATD),
conveys aromatase inhibitory and marked
tumor regression activities (—80%)^^^'
^^^.
On the
other hand, the introduction of a C^-methyl into
l,4-androstadiene-3,l7-dione as in Atamestane
(1 -methylandrosta-1,4-diene-3,17-dione, SH-489),
apparently enhances its affinity
(K^
~2 nM vs
K^
of 29 nM for 4-OHA) for the human placental aro-
matase while slowing its inactivation of the
enzyme, thereby reducing the production of estro-
genic products^ ^^' ^^^. The compound along with
its 1,2 methylene-substituted congeners has been
evaluated in Phase I clinical trials for possible
therapy of estrogen-dependent conditions such
as breast cancer and benign prostatic hypertrophy.
Additional steroidal agents explored for their
aromatase suicide inactivation include androst-
5-ene-7,l 7-dione and its 19-hydroxy derivative^^^.
Turnover-dependent irreversible inactivation of
the enzyme via protein modification is also
achieved by introducing a 6-keto group into the
steroid skeleton (Figure 7.33)^^^"^^^. Monitoring
the ^H:
^^^C
ratio in studies with the C-19 double-
labeled inhibitor indicates that the C-19 methyl,
one of the C-19 hydrogens and, from a separate
double label experiment, the Ip-hydrogen, are
retained in the covalently bound species^^^. These
findings do not define the underlying inactivation
mechanism but appear to exclude the involvement
of C-19 demethylation and aromatization,
although normal aromatization is possible because
6-oxoestrone and 6-oxoestradiol are concurrently
formed.
Exemestane, 6-methylene-androsta-1,4-diene-
3,17-dione (Figure 7.32), is an aromatase inhibitor
with an
IC^Q
for inhibition of human placental
aromatase comparable to that of 4-OHA^^^' ^^^.
The
K^
(nM) and
1^2
(min) values for the inactiva-
tion processes were 26 ± 1.4 and 29.0 ± 7.5, and
13.9 ± 0.7 and 2.1 ± 0.2, for Exemestane and
4-OHA, respectively. In spite of its relatively
slow inactivation of aromatase, Exemestane is a
more potent agent in experimental animals^ ^^, and
also much more effectively causes regression of
290
M.A. CorreJa and P.R. Ortiz de Montellano
[Fe]
HO-V
4-Hydroxyandrostenedione
(4-HOA)
Figure 7.33.
A
possible mechanism for the inactivation of
aromatase
by 4-OHA.
chemically induced mammary tumors in rat
than MDL18962 or SH-4895i^ Because of its
oral effectiveness and potent irreversible aro-
matase inhibition, Exemestane has been recently
approved and introduced into the global market
under the name Aromasin^^^^^ •.
MDL 18,962 10-(2-propynyl)estr-4-ene-3,17-
dione is one of the most potent of the mechanism-
based inactivators of aromatase, with
K^
values of
the order of 3-4 nM, and ty2 of 9 and 6 min,
respectively, for the human and baboon placental
aromatases^^^'
^^"^^ ^^^^ ^^^' ^^^
pj^ase I clinical trials
and preclinical findings indicate that MDL 18,962
is indeed effective in lowering estrogen levels^^^.
The time-dependent inactivation of aromatase
by 10-hydroperoxy-4-estrene-3,17-dione, reportedly
a mechanism-based inactivator of
aromatase^^'*'
^^^,
is inhibited by NADPH or an alternative substrate
and is partially reversed by dithiothreitol. Other
high affinity 10-substituted analogs, such as the
mechanism-based inhibitor 10-P-mercaptoestr-
4-ene-3,l7-dione, and the competitive inhibitors
(19i^)-10-oxiranyl and lO-thiiranyl-estr-4-ene-
3,17-diones and 10-p,-MeSCH2-estr-4-ene-3,17-
dione, have been reported^^i, 492,494,536,537
Q_IQ^
C-2 hydroxyethyl bridged steroids synthesized as
stable carbon analogs of the 2p-hydroxylated 19-
oxoandrostenedione, a putative intermediate in the
aromatization reaction, have been found to be
potent competitive inhibitors of
the
human placen-
tal aromatase^^^. In contrast, related halohydrin
analogs are potent mechanism-based aromatase
inhibitors^"^^.
Finally, 4p,5p-epoxyandrostenedione as well
as its 19-hydroxy and 19-oxo derivatives have
been examined as aromatase inhibitors. The epox-
ides were found to be weak competitive inhibitors,
whereas the 19-hydroxy and 19-oxo derivatives
were largely ineffective^^^.
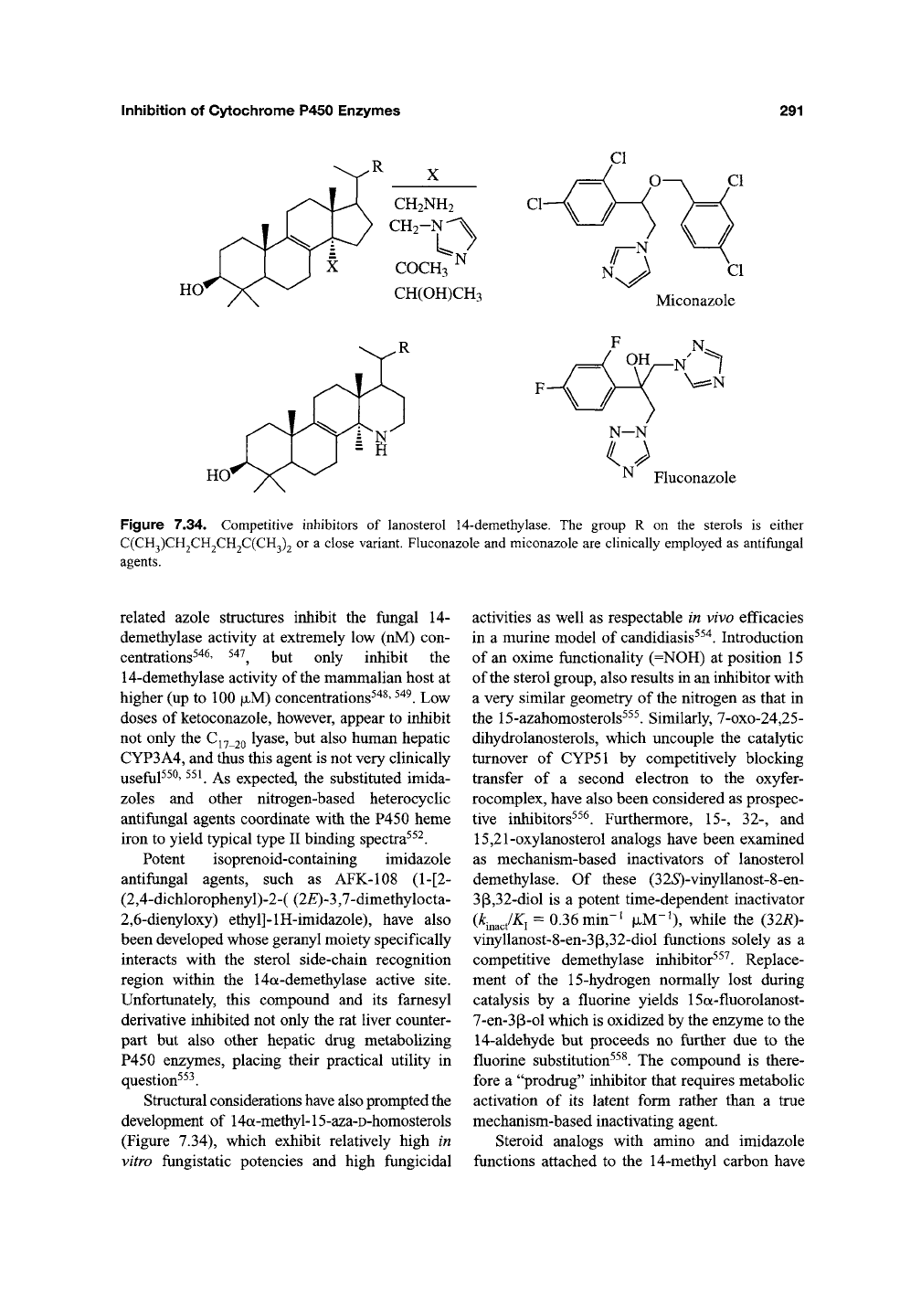
5.3. Lanosterol 14-Demethylation
The 14-demethylation of lanosterol is a
key step in the biosynthesis of cholesterol. The
preferential inhibition of the P450 enzyme that
catalyzes this reaction by a number of substituted
imidazoles, pyridines, pyrimidines, and other
lipophilic heterocycles has been exploited in the
construction of clinically important antifungal
agents^'
The antifungal action following the
inhibition of 14-demethylase is thought to result
from the accumulation of 14-methyl sterols in the
membranes of susceptible ftmgi that cause deleteri-
ous changes in their membrane permeability^"^^"^^^.
Miconazole (Figure 7.34), fluconazole
(Figure 7.34), ketoconazole (Figure 7.1), and
Inhibition
of
Cytochrome
P450
Enzymes
291
COCH3
CH(0H)CH3
CI
/=/
O-
N^
CI
Miconazole
HQ
N-N
^
Fluconazole
Figure
7.34.
Competitive inhibitors
of
lanosterol
14-demethylase.
The
group
R on the
sterols
is
either
C(CH3)CH2CH2CH2C(CH3)2
or a
close
variant.
Fluconazole
and
miconazole
are
clinically
employed
as
antifungal
agents.
related azole structures inhibit the fungal 14-
demethylase activity at extremely low (nM) con-
centrations^"*^' ^^'^^ but only inhibit the
14-demethylase activity of the mammalian host at
higher (up to 100
|JLM)
concentrations^"^^'
^^^.
Low
doses of ketoconazole, however, appear to inhibit
not only the C^^ 20 ty^se, but also human hepatic
CYP3A4, and thus this agent is not very clinically
useful^^^' ^^^ As expected, the substituted imida-
zoles and other nitrogen-based heterocyclic
antifungal agents coordinate with the P450 heme
iron to yield typical type II binding spectra^^^.
Potent isoprenoid-containing imidazole
antifungal agents, such as AFK-108 (l-[2-
(2,4-dichlorophenyl)-2-((2£)-3,7-dimethylocta-
2,6-dienyloxy) ethyl]-IH-imidazole), have also
been developed whose geranyl moiety specifically
interacts with the sterol side-chain recognition
region within the 14a-demethylase active site.
Unfortunately, this compound and its famesyl
derivative inhibited not only the rat liver counter-
part but also other hepatic drug metabolizing
P450 enzymes, placing their practical utility in
question^^^.
Structural considerations have also prompted the
development of 14a-methyl-15-aza-D-homosterols
(Figure 7.34), which exhibit relatively high in
vitro fungistatic potencies and high fungicidal
activities as well as respectable in vivo efficacies
in a murine model of candidiasis^^'^. Introduction
of an oxime functionality (=NOH) at position 15
of the sterol group, also results in an inhibitor with
a very similar geometry of the nitrogen as that in
the 15-azahomosterols^^^. Similarly, 7-oxo-24,25-
dihydrolanosterols, which uncouple the catalytic
turnover of CYP51 by competitively blocking
transfer of a second electron to the oxyfer-
rocomplex, have also been considered as prospec-
tive inhibitors^^^. Furthermore, 15-, 32-, and
15,21-oxylanosterol analogs have been examined
as mechanism-based inactivators of lanosterol
demethylase. Of these (32*S)-vinyllanost-8-en-
3p,32-diol is a potent time-dependent inactivator
(^inac/^i = 0.36 min-^
|JLM-^),
while the (32i?)-
vinyllanost-8-en-3p,32-diol functions solely as a
competitive demethylase inhibitor^^^. Replace-
ment of the 15-hydrogen normally lost during
catalysis by a fluorine yields 15a-fluorolanost-
7-en-3p-ol which is oxidized by the enzyme to the
14-aldehyde but proceeds no further due to the
fluorine substitution^^^. The compound is there-
fore a "prodrug" inhibitor that requires metabolic
activation of its latent form rather than a true
mechanism-based inactivating agent.
Steroid analogs with amino and imidazole
functions attached to the 14-methyl carbon have

292 M.A. Correia and P.R. Ortiz de Montellano
been developed as inhibitors of lanosterol 14a-
demethylation (Figure 7.34)^^^. Steroids with a
14-(l-hydroxyethyl), 14-(l-oxoethyl), or 14-car-
boxyl group are competitive inhibitors of the
enzyme (Figure 7.34)^^^"^^^. Lanosterol analogs
in which the 14a-methyl group has been replaced
by a vinyl, ethynyl, allyl, propargyl,
1-hydroxy-
propargyl,
1-ketopropargyl,
difluoromethyl,
epoxide, or episulfide moiety have been synthe-
sized as potential mechanism-based inhibitors of
lanosterol 14a-demethylase^^^~^^^. Although the
ethynyl sterols are clearly mechanism-based inac-
tivators of the enzyme^^^, most of these com-
pounds act as simple competitive inhibitors (e.g.,
the epoxide and episulfide analogs). Finally,
because of their cholesterol lowering potential,
14a-demethylase inhibitors have also been
considered as hypolipidemic agents^^^.
5.4. Other Biosynthetic
Sterol Hydroxylases
A current strategy for the development of new
drugs for the treatment of prostatic cancer is the
development of inhibitors of CYP17-dependent
androgen biosynthesis. Thus, as in the case of
other steroidogenic enzymes, an intense search
has been undertaken for reversible and irreversible
CYP17 inhibitors^^^. The resulting agents include
substituted imidazoles, pyridines, pyrimidines,
and other lipophilic heterocycles as the enabling
moiety^
Of these, abiraterone
(17-(3-
pyridyl)androsta-5,16-dien-3p-ol), a potent inhi-
bitor (IC5Q, 4 nM) of the hydroxylase activity of
human cytochrome CYP17, has been employed
clinically^^^ Its potency is apparently due to acti-
vation of the 16,17-double bond that is required
for irreversible binding of these pyridyl steroids to
CYPn^^^ Analogs such as [17-(5-
pyrimidyl)androsta-5,16-diene-3p-ol] and its 3-
acetyl derivative, which are even more potent
CYP17 inhibitors in rats than abiraterone, are par-
ticularly promising^^^. With testicular microsomal
CYP17,
these compounds apparently exhibit the
dual characteristics of a type II binding spectrum
and noncompetitive inhibition^^^
Another mechanism-based inactivator of
both the 17-hydroxylase and C-17/C-20 lyase
activities of CYP17 is 17p-(cyclopropylamino)-
androst-5-en-3p-ol^^^. This compound reportedly
does not inhibit P450g^^ or the sterol 21-hydroxy-
lase.
In contrast, sterols with a 17-difluoromethyl
group selectively inactivate the C-21 hydroxylase,
whereas sterols with a 17-dichloromethyl, -vinyl,
or -ethynyl function inactivate both the CI7- and
C21-hydroxylases^^. More recently, 20-fluoro-
17(20)-pregnenolone derivatives were designed as
enol mimics of pregnenolone. All of the targeted,
novel fluoroolefins were found to be potent
inhibitors of C-17(20) lyase579.
Similar strategies have been used to develop
inhibitors that target the C-18 hydroxylation
involved in the biosynthesis of aldosterone^^^"^^^.
Thus,
aldosterone analogs with C-18 iodomethyl,
chloromethyl, allyl, propargyl, vinyl, and methyl-
thiomethyl functionalities have been synthesized
and some have been found to irreversibly inactivate
the enzyme. An active site-directed 18-acetylenic
deoxycorticosterone [21 -hydroxy-13 (-2-)propy-
nyl)-18-nor-preg-4-ene-3,20-dione, MDL19,347]
is a promising inactivator of the rat and rhesus
monkey adrenal corticosterone 18-hydroxylase
(K^ ^ 38 nM; f,/2, 4.6 min) that also effectively
reduces plasma aldosterone levels in
vivo^^^.
This
compound appears to be a relatively selective
inhibitor of aldosterone biosynthesis, as it fails to
inhibit the
11
p-hydroxylation of corticosterone and
j)Q^586 Rationally developed inhibitors such
as these may be useful in the management of con-
ditions such as hypertension, hypokalemia, and
edema that are associated with hyperaldosteronism.
5.5. Fatty Acid and Leukotriene
Monooxygenases
Microsomal P450 enzymes of the CYP2C and
4A subfamilies in tissues such as the liver, lung,
kidney, peripheral vasculature, and polymor-
phonuclear leukocyte oxidize a variety of fatty
acids,
including arachidonic acid and its deriva-
tives,
to physiologically active metabolites.
Some of these metabolites are well recognized
as biologically important regulators of renal,
pulmonary, and cardiac function and vascular
tone^^^"^^^.
Two general classes of such vaso-
active metabolites are known to be produced in
vascular and extravascular tissues: EETs gener-
ated by CYP2C epoxygenases, and HETEs
products hydroxylated at the o)- and w-l positions
Inhibition of Cytochrome P450 Enzymes
293
by CYP4A^^^-^^^. Furthermore, with the excep-
tion of 20-HETE, all the other arachidonic acid
products occur as stereo- and regioisomers that
vary considerably in their biological activities and
potencies^92, 593
jj^^s^
55.^ 8,9-, 11,12-, and
14,15-EETs are potent vasodilators, specially in
various capillary beds, that act through activation
of K+-channels in vascular smooth muscle cells.
In contrast, 12(i^)- and 20-HETEs are potent vaso-
constrictors. 20-HETE is a particularly potent
cerebral and renal microvessel vasoconstrictor, as
well as a mediator of other important physiologi-
cal processes (reviewed in Chapter 11). The
altered production of EETs and 20-HETE in vari-
ous genetic and experimental models of patholog-
ical diseases has led to their consideration as
plausible causative factors. Thus, not surprisingly,
the P450 enzymes responsible for their biosynthe-
sis have been singled out as key targets not only
for defining the pathological roles of these
metabolites, but also as possible sites for pharma-
cological intervention in the treatment of these
conditions^^^"^^^. In this context, both the previ-
ously existing and new agents have been exploited
as reversible or irreversible inhibitors of these
enzymes.
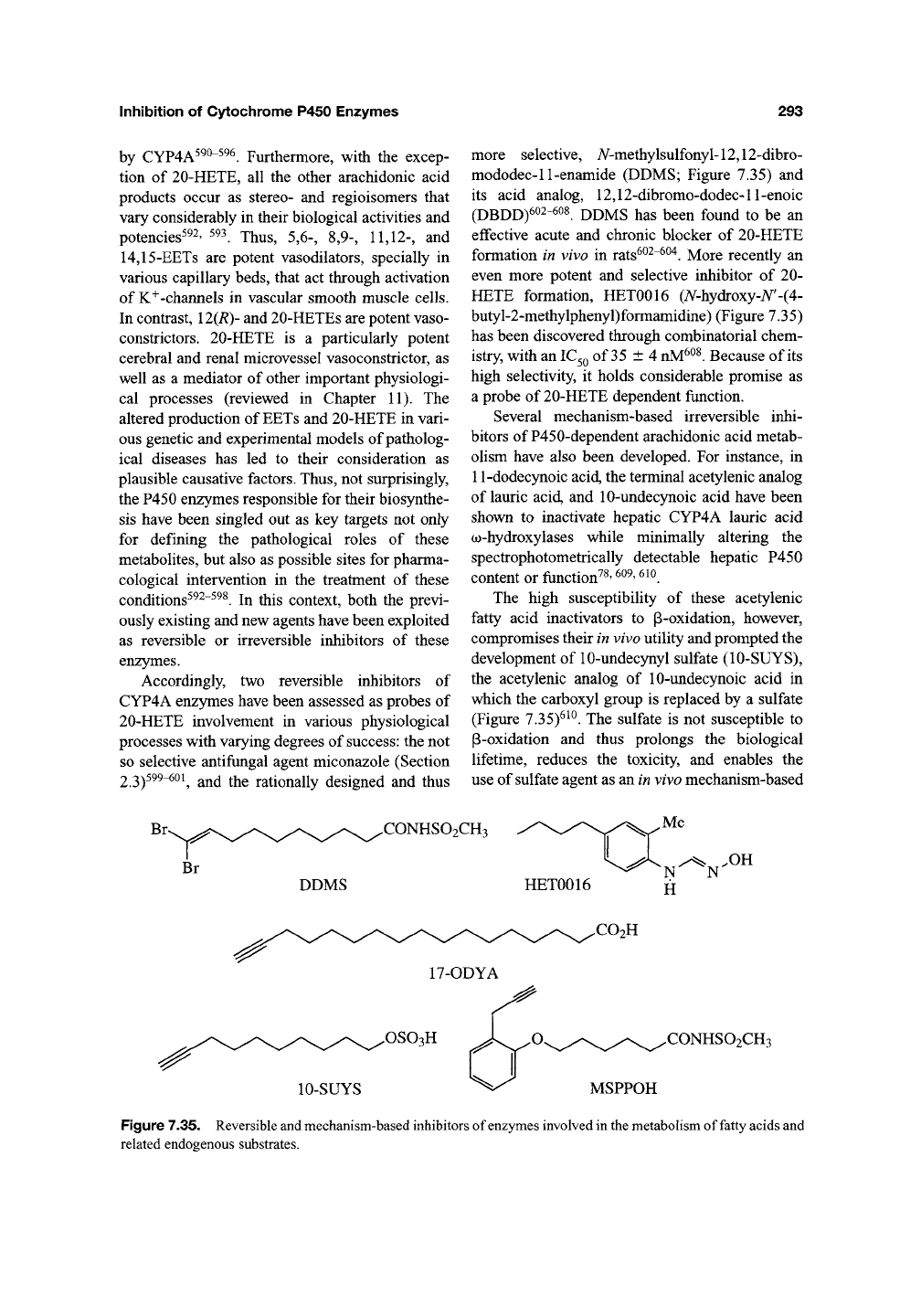
Accordingly, two reversible inhibitors of
CYP4A enzymes have been assessed as probes of
20-HETE involvement in various physiological
processes with varying degrees of success: the not
so selective antifungal agent miconazole (Section
2.3)^^^"^^^
and the rationally designed and thus
more selective, A/-methylsulfonyl-12,12-dibro-
mododec-11-enamide (DDMS; Figure 7.35) and
its acid analog, 12,12-dibromo-dodec-ll-enoic
(DBDD)602-^oi DDMS has been found to be an
effective acute and chronic blocker of 20-HETE
formation in vivo in rats^^^"^^"^. More recently an
even more potent and selective inhibitor of 20-
HETE formation, HET0016 (A^-hydroxy-A^'-(4-
butyl-2-methylphenyl)formamidine) (Figure 7.35)
has been discovered through combinatorial chem-
istry, with an
IC3Q
of 35 ±4
nM^^^.
Because of its
high selectivity, it holds considerable promise as
a probe of 20-HETE dependent function.
Several mechanism-based irreversible inhi-
bitors of P450-dependent arachidonic acid metab-
olism have also been developed. For instance, in
11-dodecynoic acid, the terminal acetylenic analog
of lauric acid, and 10-undecynoic acid have been
shown to inactivate hepatic CYP4A lauric acid
(^-hydroxylases while minimally altering the
spectrophotometrically detectable hepatic P450
content or function^^'
^°^'
^^^.
The high susceptibility of these acetylenic
fatty acid inactivators to p-oxidation, however,
compromises their in vivo utility and prompted the
development of 10-undec5niyl sulfate (10-SUYS),
the acetylenic analog of 10-undecynoic acid in
which the carboxyl group is replaced by a sulfate
(Figure 7.35)^^^. The sulfate is not susceptible to
P-oxidation and thus prolongs the biological
lifetime, reduces the toxicity, and enables the
use of sulfate agent as an in vivo mechanism-based
CONHSO2CH3
DDMS HET0016
Me
x-^ .OH
N N
H
.CO2H
17-ODYA
.OSOgH
10-SUYS
.CONHSO2CH3
MSPPOH
Figure 7.35. Reversible and mechanism-based inhibitors of enzymes involved in the metabolism of fatty acids and
related endogenous substrates.

294
M.A.
Correia
and P.R.
Ortiz
de
Montellano
CYP4A inactivator^^^. Administration of a
single dose of 10-SUYS, a potent and selective
mechanism-based CYP4A inactivator, acutely
reduced the mean arterial blood pressure as well
as the urinary 20-HETE excretion in sponta-
neously hypertensive rats, consistent with the
inactivation of renal 20-HETE formation^ ^^
These findings thus suggest that 20-HETE could
play an important role in blood pressure regulation
in hypertensive states and that the inhibition of
its synthesis in these conditions may be of thera-
peutic benefit^ ^^
CYP4A and CYP2C isoforms are also
inactivated by the acetylenic fatty acid analog,
17-octadecynoic acid (17-ODYA; Figure 7.35),
in a mechanism-based process^ ^^~^^'*. 17-ODYA
has been particularly useful in vivo to probe the
involvement of these metabolites in physiological
and/or pathological processes^^v, 598, 603, 605-609,
612-618 ji^g ygg jj^g helped elucidate the specific
role of 20-HETE in the myogenic activation and
h)^oxic dilation of skeletal muscle resistance
arteries as well as the vasodilatory effects of NO
in the renal microcirculation^^^'
^^^' ^^'^'
^^l
The acetylenic fatty acids 15-hexadecynoic
(15-HDYA) and 17-ODYA have also been
explored as modulators of leukotriene B^ (LTB^),
an important and clinically relevant inflamma-
tory mediator, and its physiologically active
(o-hydroxylated metabolite^i^'^22. Both 15-HDYA
and 17-ODYA inactivated the polymorphonuclear
leukocytic LTB^ w-hydroxylase in whole cells
and cell lysates^^^. In contrast, the shorter-chain
acid, 10-undecynoic acid was much less effective,
while the saturated analogs of 15-HDYA and
17-ODYA were inactive. 15-HDYA and 17-
ODYA also inactivate pulmonary prostaglandin
(o-hydroxylases^^^.
The mechanism-based inactivator 1-ABT
(Section 3.3.5), which is not very selective and
inactivates multiple P450 enzymes, including
those responsible for the synthesis of both EETs
and 20-HETE, has also been used, alone or in con-
junction with other inhibitors, to assess the role of
these metabolites in skeletal muscle angiogenesis
induced by electrical stimulation as well as in
the renal and vasoconstrictor actions of angio-
tensin
IP'^^'
602,
608,
624^
Mechanism-based inhibitors of EET formation
have also been used as probes of the physiological
roles of these metabolites. One such agent is
iV-methylsulfonyl-6-(2-propargyloxyphenyl)hexa-
namide (MSPPOH; Figure 7.35)^^^'
^25-627
Finally, since fatty acid hydroxylations at the
0),
(0-1, (0-2, (0-3, and (o-4 positions also occur in
plants and bacteria^^^'
^^^,
acetylenic fatty acids
have also been employed to examine whether
these hydroxylases are mechanistically similar
to their mammalian counterparts and/or to iden-
tity the role of specific plant and bacterial P450
enzymes^^^' ^^^. Thus, midchain and terminal
acetylenes such as 10-dodecynoic acid have been
used as probes of plant lauric acid (o—hydroxy-
lases^^'6^^.
Similarly, 17-ODYA has been shown to
inactivate
P450BJ^.3
(CYP108), an enzyme that
hydroxylates fatty acids at positions other than the
terminal ((o) carbon, through a heme alkylation
mechanism^^o.
6. Summary
Our knowledge of the structure and function
of P450 enzymes has greatly expanded over the
past three decades, as has our appreciation of
the chemical functionalities that they accept as
substrates and of the moieties that can inhibit or
terminate their catalytic action. As a result, multi-
ple routes are now available for the inhibition or
inactivation of P450 enzymes that target either the
protein and/or the heme. This knowledge has been
exploited in the design and construction of both
reversible and irreversible inhibitors of increasing
potency, specificity, and potential practical utility.
One area of continuing growth is that of reversible
inhibitors that, by simultaneously conforming to
the predominantly lipophilic contours of the P450
active site and providing a nitrogen that coordi-
nates to the heme iron atom, have achieved
enhanced potency and specificity. These agents
include the clinically relevant azole antifungal and
cancer chemotherapeutic drugs (Section 5).
The high promise of irreversible P450
inactivating agents has not yet culminated in
agents of large-scale practical utility. Progress
in the field has continued to provide instruction on
approaches for the design of mechanism-based
inhibitors that inactivate a P450 enzyme with high
specificity and negligible release of reactive
metabolites into the medium, whether the inacti-
vation involves modification of the protein or the