Патофизиология обмена веществ
Подождите немного. Документ загружается.

или в качестве инактиваторов Т-супрессоров (ретровирусы) либо стимуляторов Т-
эффекторов. В этом случае аутоаллергический процесс может быть следствием вирус-
индуцированного дефицита супрессоров и/или избытка эффекторов. В то же время
иммунологический цитолиз присущ течению инфекций у наследственно
предрасположенных субъектов.
Провоцирующая роль вирусов в генезе аутоиммунного цитолиза осуществляется
через интерлейкины и интерфероны, особенно -интерферон, при вирусном поражении
поджелудочной железы. Эти цитокины вызывают экспрессию антигенов ГКГС на -
клетках и аутопрезентацию поверхностных антигенов -клеток к последующему
аутоиммунному цитолизу, а также появлению неоантигенов при персистирующих
вирусных поражениях.
К химическим диабетогенам относятся аллоксан, мочевая кислота, стрептозоцин,
дитизон, вакор (средства для борьбы с грызунами), бычий сывороточный альбумин
(входит в состав коровьего молока), нитрозамины и нитрозомочевина (содержатся в
копчёных продуктах), пентамидин (средство для лечения пневмоцистоза), продукты,
содержащие пищевые цианиды (абрикосовое зерно, миндаль, африканский корнеплод
кассава, которым питается около 400 млн. аборигенов, и др.). Курение и алкоголь
способствуют повышению уровня цианидов в крови, усиливают проявления
аутоиммунитета, способствуют развитию гемохроматоза и панкреатита.
В противовес диабетогенам описаны вещества с протекторным эффектом, так
называемые антидиабетогены. Среди них называют серосодержащие аминокислоты,
дефицит которых повышает токсичность пищевых цианидов, антиоксиданты, цинк
(участвует в депонировании инсулина), витамин РР (тормозит процессы апоптоза и
некроза, используется для лечения ИЗСД), полиненасыщенные жирные кислоты из
морепродуктов (подавляют синтез широко известных ИЛ-1 и ФНО-α).
Основные механизмы химического повреждения панкреатических островков – это
интерлейкин-зависимая экспрессия отсутствующих в норме на мембране -клеток DR-
белков, аутоиммунная альтерация и аутоаллергия, вызванная перекрёстными или
общими антигенными детерминантами, а также иммунный ответ на экспрессию
неоантигенов, обусловленный деструкцией -клеток. В то же время возможно подавление
пролиферации -клеток антиклеточными антителами и медиаторами аутоиммунного
воспаления.
Подводя итог сказанному относительно иммунных процессов ИЗСД, выделим
главные. Это, во-первых, аллергический инсулит, вызванный цитотоксическими Т-
лимфоцитами (клеточно-опосредованный тип аллергии) вследствие экспрессии на
мембране -клеток отсутствующих в норме DR-белков. Не исключена экспрессия
неоантигенов – продуктов латентного вирусного генома, а также аномальная экспрессия
генов ГКГС второго класса на -клетках. Во-вторых, гуморально-опосредованный тип
деструкции -клеток, который представлен комплемент-зависимой и антитело-
опосредованной клеточной цитотоксичностью (цитотоксический, или цитолитический, тип
аллергических реакций). Выделяемые цитокины (ИЛ-1, ФНО-a, лимфотоксин, -
интерферон, фактор активации тромбоцитов, простагландины) ещё до выраженной
аутоиммунной деструкции -клеток приводят к торможению секреции инсулина.
Особенно это относится к ИЛ-1, который снижает чувствительность -клеток к глюкозе.
Указанные цитокины, выделяемые лимфоцитами и макрофагами, обладают
цитотоксическим, антипролиферативным и антисекреторным эффектами. Помимо
аутоаллергического цитолиза, для ИЗСД характерно выключение митотической
активности -клеток.
Патогенез ИЗСД. Ключевое звено патогенеза ИЗСД – прогрессирующая гибель -
клеток панкреатических островков. Это приводит к изменению гетероклеточных
взаимоотношений в островках, инсулинопении, избытку островковых и внеостровковых
контринсулярных гормонов. В результате нарушаются утилизация глюкозы и все виды
71
метаболизма. Хронические нарушения метаболизма порождают осложнения ИЗСД,
главные из которых связаны с ангиопатиями.
Роль провоцирующего вирусного и/или химического диабетогена состоит в
индукции аутоиммунной альтерации. У 10 % больных с подтипом ИЗСД 1b (в сочетании с
системной аутоиммунной полиэндокринопатией) провокация не является необходимой. У
больных с подтипом ИЗСД 1а провоцирующее событие должно произойти в раннем
онтогенезе или даже до рождения, т.к. ИЗСД – заболевание с длительным
иммунологическим продромом и периодом метаболической компенсации. Интервал от
дебюта аутоиммунного процесса до начала интолерантности к глюкозе составляет 3-4
года, а наиболее длительный период между первыми проявлениями снижения способности
к выработке инсулина и явной метаболической декомпенсации – 1-12 лет. Пик
заболеваемости ИЗСД приходится на возрастные периоды от рождения до 3 и от 9 до 13
лет. После 14 лет потенциальные возможности эндогенных диабетогенов спровоцировать
деструкцию -клеток снижаются.
Морфофункциональная основа ИЗСД. В ответ на иммунологическую альтерацию в
панкреатических островках развивается инсулит, проявляющийся гибелью -клеток,
экссудативными изменениями, инфильтрацией островков лимфоцитами, макрофагами,
эозинофилами, извращением нейроваскулярных взаимоотношений, нарушениями
топографии клеток и межклеточных контактов. К моменту формирования клинически
явного диабета вес поджелудочной железы уменьшается в два, масса островков – в три
раза, а В-клеток – более чем в 850 раз. В то же время в дезорганизованных островках
растет доля А-клеток (до 75 %) и δ-клеток (до 25 %). В результате соотношение глюкагон/
инсулин в крови больных ИЗСД по мере развития болезни стремится к бесконечности.
Классификация сахарного диабета. Первичный сахарный диабет I типа [синонимы:
инсулинозависимый, гипоинсулинемический, юношеский (ювенильный) ИЗСД)]
составляет 20 % от общего числа случаев первичного сахарного диабета. Подтипы: Iа –
обусловлен комбинацией генетического и средового воздействия; Ib – первичный,
генетически обусловленный без экзогенной провокации; Iс – с первичным поражением -
клеток экзогенными химическими и вирусными диабетогенами.
Первичный СД II типа (инсулинонезависимый, гиперинсулинемический, взрослых,
пожилого возраста, тучных, ИНСД) составляет 80 % всех случаев больных СД со
следующими подтипами:
IIа – ИНСД у нетучных больных;
IIb – ИНСД у тучных больных;
IIс – ИНСД юношеского возраста.
Термины «ИЗСД», «ИНСД» описывают особенности клинического течения
(склонный к кетоацидозу и резистентный к кетоацидозу, Таблица 3.1), а термины «I и II
типы» относят к патогенетическим механизмам болезни (результат доминирования
аутоиммунного или иных механизмов).
Вторичный СД (это гипергликемические, или диабетические синдромы, которые
являются следствием болезней, поражающих поджелудочную железу или систему
регуляции углеводного метаболизма).
Вторичный диабет вызванный неаутоиммунной деструкцией -клеток (хронический
панкреатит, рак, гемохроматоз, кистоз, травмы);
вторичный диабет, вызванный эндокринными расстройствами с гиперпродукцией
контринсулярных гормонов (синдром Кушинга, акромегалия, феохромоцитома,
глюкагонома, гипертиреоидизм, гиперплазия эпифиза);
вторичный ятрогенный диабет в результате применения медикаментов
(кортикостероиды, АКТГ, оральные контрацептивы, пропранолол, антидепрессанты,
некоторые мочегонные);
вторичный диабет при генетически детерминированных синдромах
(липодистрофии, гипоталамические формы вторичного ожирения, гликогеноз I типа,
болезни Дауна, Шерешевского, Клайнфельтера.
72

Таблица 3.1
Критерии отличия ИЗСД и ИНСД
Абсолютный дефицит инсулина Относительный дефицит инсулина
Аутоиммунный процесс против -клеток Аутоиммунный процесс отсутствует
Отсутствие первичной инсулинорезистентности Первичная инсулинорезистентность
Высокий риск кетоацидоза Низкий риск кетоацидоза
Ссвязи с тучностью не прослеживается Прослеживается связь с тучностью
Конкордантность однояйцовых близнецов 30-
50%
Конкордантность однояйцовых
близнецов 90-100%
Еще раз подчеркнем, что ключевым звеном патогенеза ИЗСД является
прогрессирующая гибель -клеток вследствие аутоиммунной альтерации. Определены
антигенные маркёры ИЗСД – это антигены ГКГС DR
3
, DR
4
, DQ
3.2
.
В семьях, где отец болен ИЗСД, число больных детей в 4-5 раз больше, чем в семьях,
где больна мать.
Иммунологический конфликт матери и плода по системе АВ0 и Rh
+
увеличивает риск
развития ИЗСД.
Однако генетическая предрасположенность лишь создает высокую вероятность
заболевания. Для реализации необходимы инфекционные и неинфекционные
диабетогенные факторы. Механизм действия диабетогенов связан с интерлейкин-
зависимой экспрессией аутоантигенов -клеток. Имеются основания предполагать, что
значительная часть больных ИНСД – это лица, находящиеся в ранней стадии эволюции
СД, но еще имеющие достаточно инсулина, чтобы предотвратить кетоацидоз. ИНСД у
тучных имеет существенный патогенетический механизм – продукция адипоцитами
контринсулярного цитокина ФНО-a. ИЗСД и ИНСД имеют множество патогенетических
звеньев, в то же время нельзя отрицать существование смешанных и переходных форм.
ИНСУЛИНОНЕЗАВИСИМЫЙ САХАРНЫЙ ДИАБЕТ
ИНСД – это гетерогенная по этиологии и патогенезу группа заболеваний,
характеризующаяся мультифакториальной наследственной предрасположенностью,
относительной инсулиновой недостаточностью и инсулинорезистентностью. До сих пор
дебатируется вопрос о том, что первично в патогенетической основе ИНСД – длительное
действие выявляющих его факторов или возрастная экспрессия предрасполагающих генов.
Поскольку доказано, что у большей части больных инсулинонезависимый сахарный
диабет сочетается с ожирением и коррелирует с пожилым возрастом, значительная их
часть имеет характерную комбинацию расстройств, объединённых в единый синдром –
метаболический Х-синдром. К нему относят ГЛП IV или V типов, ускоренное развитие
атеросклероза, ожирение андроидного типа, стеатоз печени, гипертензию, гиперурикемию,
нефропатию.
Семейный риск ИНСД значителен – до 40 % сибсов и треть потомства у больных
имеет толерантность к глюкозе. В отличие от ИЗСД, для ИНСД выявлены регионы, где
проявляются эффект родоначальника и последствия инбридинга (остров Науру, где СД
больны 83 % коренных жителей и район расселения индейцев пима, в Северной Америке,
среди которых до 86 % больных СД). Несмотря на это, лишь для отдельных моногенных
менделирующих разновидностей ИНСД установлен точный тип наследования.
Основные патогенетические звенья ИНСД представлены следующими дефектами:
недостаточная секреция инсулина для утилизации глюкозы, особенно в течение
первого часа после поступления углеводов, из-за снижения чувствительности
глюкорецепторов В-клеток островков к глюкозо-стимулу;
инсулинорезистентность вследствие аномалий молекулы инсулина, связывания
инсулина циркулирующими в крови антителами к нему, наличием антител к
инсулиновым рецепторам в клетках, уменьшением их числа и т.п.;
73
усиленным образованием глюкозы печенью на протяжении полных суток, тогда как в
норме синтез глюкозы происходит только днем.
недостаточный ответ -клеток на глюкозный сигнал без их деструкции,
синтез «неправильного» проинсулина, либо нарушение его протеолиза,
избыток циркулирующих антагонистов инсулина,
аномалии инсулиновых рецепторов,
блокада инсулиновых рецепторов,
пострецепторный блок на разных уровнях в клетках-мишенях.
Все эти дефекты, вероятно, генетически детерминированы, причем большинство из
них – моногенно. В отдельности подобные механизмы встречаются редко, в сумме эта
гетерогенная группа составляет большую часть больных с диагнозом ИНСД. В
доказательство высказанного приводим примеры случаев ИНСД при генетически
обусловленных отдельных дефектах, составляющих комплекс патогенетических
механизмов и ИНСД. Так, при диабете взрослых в юности (один из подтипов ИНСД)
выявляется наследственный дефект фермента глюкокиназы (ген в коротком плече
хромосомы 7). Этот фермент обусловливает чувствительность -клеток и гепатоцитов к
уровню глюкозы. Как следствие, данный дефект вызывает относительную «глухоту» -
клеток к глюкозному стимулу. Другим характерным проявлением ИНСД является скорее
нарушение синтеза гликогена (гликогенез), чем захват глюкозы клетками и её окисление.
Этот дефект наблюдается даже у нетучных родственников многих больных ИНСД. Это
указывает на взаимосвязь с аллелью гликогенсинтетазы А
2
и высокой вероятностью
развития ИНСД (30 % частоты против 8 % в контрольной группе).
Аномалии самой молекулы инсулина могут возникать в результате мутации его
структурного гена, кодирующего синтез биологически дефектных молекул этого гормона.
Описаны единичные случаи ИНСД, обусловленного неполным превращением
проинсулина в инсулин в ходе протеолиза, происходящего в грануле -клетки. В норме
только около 5 % секреторного продукта -клеток представлено проинсулином, а
биологическая активность последнего много меньше, чем инсулина. Больные такой
формой ИНСД имеют дефект структурного гена проинсулина в точке отщепления
терминального С-пептида. Это препятствует превращению проинсулина в инсулин и
терминальный С-пептид. В крови таких больных радиоиммунологическим методом
выявляется гиперинсулинемическое состояние.
Признаётся участие в патогенезе ИНСД генов-транспортёров глюкозы, в частности
GluT-2, контролирующих поступление глюкозы в гепатоциты и панкреатоциты, или GluT-
4 – в липоциты и миоциты. У некоторых пациентов ИНСД имеется избыточная экспрессия
гена Rad, участвующего в работе по системе внутриклеточной передачи гормональных
сигналов. Его продукт ингибирует инсулинозависимую активность транспортера глюкозы
GluT-4.
Велика роль генетических аномалий возможного контринсулярного фактора -
клеток – амилина, или известного под другим названием амилоидогенного пептида. В
норме амилин представляет собой пептид из 37 аминокислот и секретируется в небольших
количествах вместе с инсулином. У многих больных ИНСД избыток амилина
откладывается в синусоидных пространствах вокруг -клеток, формируя в последующем
амилоидные отложения, а синтез амилина растет параллельно секреции инсулина, что
снижает ответ -клеток на глюкозу и в последующем может вызвать ограничение их
секреторных возможностей.
У некоторых больных ИНСД обнаружены особенности гена (хромосома 19),
контролирующие пептиды инсулинового рецептора в клетках-мишенях. Так, аномальный
пептид инсулинового рецептора может служить маркером ИНСД до четверти случаев, а
тучные больные имеют низкую тирозинкиназную активность в клетках-мишенях
инсулина. У 15 % больных имеется генетическая вариация в пострецепторном блоке
клеток-мишеней, например, в структуре внутриклеточного инсулинового посредника IRS-
1. У других больных описан избыточный синтез a
2
-SH-гликопротеина, который блокирует
74
тирозинкиназную активность инсулиновых рецепторов клеток-мишеней. Выделен
мембранный гликопротеин, ингибирующий тирозинкиназную активность рецепторов
инсулина. Таким образом, чаще всего множественные пострецепторные дефекты могут
вызывать первичную резистентность -клеток к глюкозе, а клеток-мишеней инсулина – к
этому гормону.
Экзогенным фактором, выявляющим генетические аномалии при ИНСД, служит
переедание. Переедание повышает нагрузку на систему инсулиновой регуляции
метаболизма. Формирующееся при этом ожирение вызывает включение дополнительных
механизмов инсулинорезистентности. Низкая эффективность инсулина, характерная для
ИНСД, теоретически могла бы компенсироваться гиперфункцией -клеток. Однако при
данном заболевании имеетместо пониженная их чувсивительность к глюкозе, в основе
которой лежат разнообразные причины.
Для ИНСД характерны следующие особенности морфологии и функции островковых
клеток:
увеличение общего объема островковой ткани за счет возрастания числа инсулин-
продуцирующих -клеток,
нередко амилоидоз и фиброз островков,
резко уменьшено количество соматостатин-продуцирующих δ-клеток, что не может
оказывать адекватного тормозного паракринного влияния на глюкагон-продуцирующие А-
клетки,
базальная секреция инсулина при ИНСД, как минимум, нормальна.
Патогенез ИНСД. Как уже упоминалось, в патогенезе ИНСД признается
представление о первичности инсулинорезистентности и нарушении отвечаемости -
клеток на глюкозо-стимул. Это ведет к трехфазной картине течения ИНСД:
стадия начальной инсулинорезистентности и компенсации гликемии.
стадия выраженной инсулинрезистентности и относительной инсулиновой
недостаточности, интолерантности к глюкозе.
стадия снижения инсулиновой секреции и явного диабета.
Формирующаяся у больных прогрессирующая тучность усиливает
инсулинрезистентность. Для ИНСД кетоацидоз не характерен по тем же причинам, по
которым имеется ожирение – вследствие наличия инсулина, который сдерживает липолиз
и создает условия для утилизации ацетил-КоА по пути стероидогенеза и липогенеза.
Содержание малонил-КоА остается высоким, угнетая окисление жирных кислот и
перекрывая путь кетогенеза. Метаболической платой за это служат тенденции к ожирению
и крайне высокой гиперхолестеринемии. У больных ИНСД гиперхолестеринемия более
умеренная.
Патогенез усиления инсулинрезистентности по мере ожирения (особенно
гипертрофический и андроидный типы ожирения) обусловлен:
уменьшаением плотности рецепторов инсулина на поверхности жировых клеток, что
затрудняет гормон-рецепторное взаимодействие (ограничение экспрессии инсулиновых
рецепторов).
Торможение выделения адипоцитами цитокина, обладающего способностью снижать
киназную активность рецептора инсулина в миоцитах и липоцитах – ФНО-a
Биологический смысл этого состоит в ограничении притока в адипоциды субстрата
для липогенеза, тем более что названный цитокин является аппетит-подавляющим
регулятором, откуда его второе название – кахексин. Но у тучных лиц кахексиновый
механизм подавления неэффективен. В то же время ФНО-a вызывает дополнительную
инсулинрезистентность, а также цитотоксичен для -клеток, что открывает
патогенетические пути перехода ИНСД во вторую и третью фазы развития. Однако
ожирение не является единственно значимым фактором инсулинрезистентности. Важной
причиной инсулинрезистентности могут быть адаптивное снижение экспрессии
инсулиновых рецепторов при «сытом» состоянии клеток, а также иммунологические
реакции.
75
Перечисленные механизмы ответственны за прогрессирование болезни. В третьей
стадии ИНСД нет нарастания инсулинорезистентности, но уменьшаются секреторные
возможности -клеток. Опыты с удалением панкреас показывают, что 10 % массы
интактных -клеток достаточно для поддержания нормогликемии без соблюдения диеты.
Предполагается, что инсулинорезистентность приобретает при ИНСД решающее значение
только при нарушении отвечаемости -клеток на глюкозу вследствие их дефекта. По этой
причине подавляющее большинство лиц, страдающих тучностью и периферической
инсулинрезистентностью, имеют гиперинсулинемию, но не гипергликемию. При наличии
инсулинрезистентности компенсаторно развивается состояние гиперинсулинизма. Однако
для стимуляции дефектных -клеток требуется высокое содержание глюкозы,
превосходящие почечный порог. Из-за глюкозурии гипергликемия не может
увеличиваться бесконечно. При гипергликемии в 250 мг/дл и выше глюкоза теряется, а
секреторные резервы -клеток не могут быть отмобилизированы полностью. Состояние
патологического равновесия достигается при больших или меньших концентрациях
инсулина в зависимости от преобладания нсулинорезистентности или дефекта -клеток, но
инсулина всегда недостаточно по отношению к имеющемуся избытку глюкозы в крови.
Тяжелый кетоацидоз, который не купируется инсулином, связывают с
нсулинорезистентностью. Даже «малые дозы» инсулина могут вызвать повышение его
содержание в крови в 4-15 раз, не оказывая терапевтического эффекта. Это послужило
основанием для заключения, что нсулинорезистентность в условиях кетоацидоза
обусловлена избытком протонов и СЖК.
ПАТОГЕНЕЗ ОСЛОЖНЕНИЙ САХАРНОГО ДИАБЕТА
К острым осложнениям относятся коматозные состояния, к хроническим –
микроангиопатии, макроангиопатии (МиП и МаП), инсулинорезистентность, нейропатия,
нефропатия, иммунодефициты. Для ИНСД более характерны гиперосмолярная и
гиперлактацидемическая комы.
МаП встречаются чаще и проявляются хронической ишемической болезнью сердца,
нарушением мозгового кровообращения и облитерирующим атеросклерозом артерий
нижних конечностей. В патогенезе МаП ведущее значение имеет ускоренное развитие
атеросклероза, тогда как при МиП – гипергликемия. Механизмы ускоренного развития
атеросклероза множественны – гиперлипопротеинемия, гипертензия, гипергликемия,
гиперинсулинизм, тромбофилитический синдром.
Патогенез острых осложнений сахарного диабета. Кетоацидотическая кома.
По мере развития СД все пути использования избытка ацетил-КоА блокируются, за
исключением тех, которые ведут к кетозу и синтезу холестерина, метаболическому
ацидозу, потере воды и электролитов, гемоконцентрации, недостаточности
периферического кровообращения, аритмиям, шоку. Развивается компенсаторный
метаболический ацидоз с потерей натрия мочой и компенсаторным выходом из клеток
протона, что усугубляет ацидоз. Вследствие глубокой гипоксии ЦНС функции
пневмотаксического центра замещаются гаспинг-центром, развивается дыхание
Куссмауля, гипервентиляция, гипокапния, гипобикарбонатемия, что углубляет ацидоз.
Вследствие гипоксии в ткани мозга накапливается избыток лактата, что ведет к
усугублению ацидоза. Ацидоз по типу порочного круга при диабетической коме вызывает
усиление инсулинорезистентности, так как инсулин в кислой среде теряет сродство к
своему рецептору. Кроме того, инсулинорезистентность обусловлена высоким уровнем
СЖК и высвобождением контринсулярных гормонов – антагонистов инсулина
(адреналина, глюкокортикоидов, глюкагона, вазопрессина). Диабетическая
(кетонемическая, ацидотическая) кома обусловлена токсическим влиянием кетоновых тел
и тканевой гипоксии на клетки ЦНС, обезвоживанием, ацидозом. Усиленный катаболизм
белка приводит к увеличению содержания аммиака и мочевины, продукционной
гиперазотемии, что углубляет интоксикацию и гипоксию мозга. Гипоксия нейронов
76
приводит к расстройству дыхания, сосудистому коллапсу, снижению мышечного тонуса,
нарушению ВНД.
Лактоацидоз и гиперлактацидемическая кома. Встречаются довольно часто
(токсические дистрофии, циррозы печени), при сердечной недостаточности и других
болезнях и нередко в тяжёлой форме – при декомпенсации ИНСД, который лечили
бигуанидами – блокаторами глюконеогенеза.
В крови повышается уровень лактата более 5 ммоль/л при норме до 1,5 ммоль/л,
значение рН артериальной крови 7,25 ед. и менее. Лактоацидоз является результатом
гипоксии и физического переутомления. Клинически характерны дыхание Куссмауля,
гипотензия, гипотермия, обезвоживание, ацидоз, циркуляторный коллапс, отсутствие
кетонурии.
Гипергликемическая (гиперосмолярная) кома встречается реже кетоацидотической
в основном у больных старше 50 лет, чаще беспомощных. Провоцируется дегидратацией
организма (рвота, понос, лечение диуретиками, ограничение приема жидкости).
Кетоацидоз отсутствует, гипергликемия может нарастать растянуто во времени до
высоких цифр (55 ммоль/л и более). В патогенезе имеют значение следующие факторы:
Гипергликемия 55-200 ммоль/л (1000-3600 мг/дл).
Гипернатриемия, гиперхлоремия (обусловленные гиперальдостеронизмом в ответ на
дегидратационную гиповолемию),
Гиперазотемия (за счет мочевины) из-за ограничения диуреза.
Отсутствие дыхания Куссмауля, запаха ацетона.
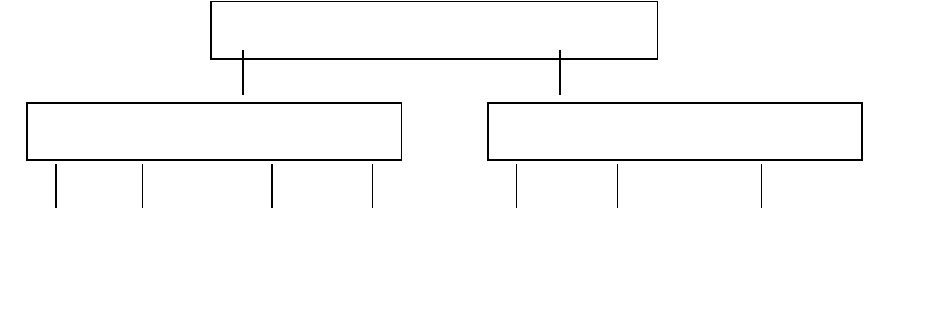
Патогенез хронических осложнений. Диабетические ангиопатии являются
главными осложнениями СД, инвалидизации и смерти больных (Схема 3.1). Понятие
«ангиопатии» включает микроангиопатии (поражение капилляров, венул, артериол,
прежде всего, их базальной мембраны) и макроангиопатии (поражение крупных артерий).
В течение СД любого типа наблюдаются комбинированная ангиопатия с
преобладанием у молодых лиц ИЗСД типа микроангиопатии, у лиц старше 40 лет и ИНСД
типа макроангиопатия с прогрессирующим развитием атеросклероза. Общим для МиП
всех локализаций являются аневризмы капилляров, утолщение стенок артериол,
капилляров, венул за счет накопления в базальной мембране гомогенных или слоистых
веществ, пролиферация эндотелия в просвет сосудов (вплоть до полной облитерация),
тучноклеточная реакция в периваскулярной ткани. Так, например, ИЗСД – главная
причина слепоты и одна из ведущих системных причин ХПН.
Схема 3.1
Патогенез микроангиопатий. В патогенезе ангиопатий имеет значение, во-первых,
неферментативное гликозилирование различных внеклеточных белков (белки базальной
мембраны гломерулярного аппарата почек, хрусталика глаза, жидкости стекловидного
тела глаза и др.). Во вторых, ферментативное превращение глюкозы в сорбитол и далее во
фруктозу внутри определенных клеток. Конечные продукты необратимого глубокого
гликозирования оказывают патогенные эффекты на соединительную ткань:
77
Диабетическая ангиопатия
Микроангиопатии Макроангиопатии
Нефро-
патия
Ретино-
патия
Нейро-
патия
Пангипопи-
туитаризм
ИБС
Инсульт
Периферический
артериолосклероз
и атеросклероз
вызывают сшивку коллагена базальных мембран микрососудов и белков плазмы,
«пришивают» липопротеиды к коллагену крупных сосудов, препятствуя работе
дренажных механизмов и ускоряя атерогенез,
делают белки депозитов устойчивыми к протеолизу,
препятствуют взаимодействию коллагена и протеогликанов, в результате чего
базальная мембрана повышает свою проницаемость,
стимулируют выделение тканями эндотелина-1, цитокинов и других факторов,
усиливающих коагуляцию, тромбообразование, спазм сосудов,
могут менять антигенные свойства белков и провоцировать иммунокомплексный
процесс.
ГЛИКОГЕНОЗЫ (БОЛЕЗНИ НАКОПЛЕНИЯ ГЛИКОГЕНА)
Гликогенозы – группа наследственных нарушений (болезней) накопления гликогена
и/или его утилизации, создающих недостаточность на путях поддержания уровня глюкозы
в крови и/или генерации энергии (мышцы). Гликогеноз (III типа) был первой
наследственной болезнью печени, для которой удалось раскрыть природу первичного
энзиматического дефекта. По традиции гликогенозы имеют номерную классификацию,
однако в настоящее время доминирует патогенетическое деление гликогенозов на
печеночные, мышечные и смешанные формы. Поражение может сопровождаться
количественными нарушениями гликогена или ограничиваться качественными его
аномалиями.
Печеночные гликогенозы поражают метаболический путь (катаболизм углеводов),
при котором нарушен гликогенолиз и вследствие этого поддержание уровня глюкозы в
крови. В то же время гликогенез не нарушен. К этой группе относят болезни Гирке,
Форбса-Кори, Херса и др. Гликогеноз I типа (гепаторенальный тип, болезнь Гирке, дефект
глюкозо-6-фосфатозы). Наследуется аутосомно-рецессивно, встречается чаще других
гликогенозов. Патогенез связывают с неспособностью организма больного превратить
субстрат глюкозо-6-фосфат, который в избытке присутствует в гепатоцитах и нефроцитах,
в глюкозу, что ведет к развитию гипогликемии, а также ацетонемии и ацетонурии,
гиперхолестеринемии, гиперурикемии. В тяжёлых случаях возможны судороги. Это
обусловливает относительный гипоинсулинизм, результатом чего является усиление
липолиза и гиперлипопротеинемия I или V типов.
У болеющих детей находят гепатомегалию (уже в период новорожденности),
задержку роста, короткое туловище, большой живот. В печени развивается стеатоз. Почки
также увеличены и содержат депозиты гликогена. В постпубертатном периоде выражены
клинические осложнения гиперурикемии (подагра, уролитиаз). Для больных характерны
гипогликемия натощак, усиливающаяся после голодания, (ограниченный подъем сахара в
крови после инъекций глюкагона и адреналина), редуцированные сахарные кривые.
Разновидностью гликогеноза I является гликогеноз Ib, к счастью, встречается не так
часто. Представляет собой редкую аутосомно-рецессивную генокопию предыдущего
расстройства, но с поражением гена транслоказы глюкозо-6-фосфата в
эндоплазматическом ретикулуме. Клиника болезни усугубляется присоединением к
классической болезни Гирке еще и нейтропении с рецидивирующими бактериальными
инфекциями (иммунодефицит). Поэтому расщепления эфира глюкозы недостаточно,
несмотря на высокую активность фосфатазы.
Другой печеночный гликогеноз III типа (болезнь Форбса-Кори). Аутосомно-
доминантный дефект фермента, катализирующего гидролиз 1-6 связей в молекуле
гликогена. Составляет четверть всех случаев гликогенозов, протекающих с
гепатомегалией. Гликогенолиз возможен в незначительных количествах, так как
фосфорилироваться могут только молекулы глюкозы, взятые из коротких цепей.
Отличается доброкачественным течением, не угрожает жизни ребенка, так как в крови нет
апактоацидоза и гиперурикемии. Гипогликемия присутствует натощак, сохраняется ответ
на глюкагон на фоне потребления углеводов, но не натощак. Гликогенез VI а типа
78
(болезнь Херса, дефицит фосфорилазы). Клиника сходна с течением гликогеноза I типа.
Встречается крайне редко.
К мышечным гликогенозам относят дефекты мышечной фосфорилазы,
фосфофруктокиназы, которые обуславливают нарушения в энергоснабжении скелетной
мускулатуры. Выявляются чаще всего при физической нагрузке и находят выражение в
мышечной слабости, миалгии, миоглобинурии, сопровождаются гемолитическими
анемиями, провоцируемыми окислителями (гипогликемия не обнаруживается). Примерои
служит: гликогеноз V типа (болезнь Мак-Ардля).
Смешанные формы гликогенозов. Гликогеноз II типа – это генерализованный
гликогеноз, поражающий все гликогенсодержащие клетки. Синонимы: гликогенная
кардиомегалия, болезнь Помпэ, дефект лизосомальной глюкозидазы. Составляет 10 % от
всех гликогенозов. Эта форма очень злокачественная, больные умирают в грудном
возрасте. Ведущий признак – кардиомегалия в грудном возрасте с развитием тяжелой
сердечной недостаточности, миалгия, увеличение языка. Гипогликемии нет, так как
печеночный гликогенолиз не страдает.
ТЕСТОВЫЕ ЗАДАНИЯ.
79
1. Выберите правильное утверждение. К группам
причинных факторов, вызывающих нарушение обмена
белков, относят:
А. Заболевания слизистой оболочки желудочно-
кишечного тракта.
Б. Заболевания крупных пищеварительных желез.
В. Голодание.
Г. Нефриты.
Д. Желтухи.
Е. Ожирение.
2. Выберите правильное утверждение. Развитие
отрицательного азотистого баланса может быть
связано со следующими патологическими
состояниями:
А. Голодание.
Б. Беременность.
В. Рост организма.
Г. Период после голодания.
Д. Протеинурия.
3. Выберите правильное утверждение. Развитие
отрицательного азотистого баланса может быть
связано со следующими патологическими
состояниями.
А. Инфекционные заболевания.
Б. Избыточная продукция анаболических
гормонов (соматотропин, андрогены).
В. Избыточная продукция катаболических
гормонов (тироксин, кортизол).
Г. Протеинурия.
4. Выберите правильное утверждение. Развитие
положительного азотистого баланса может быть
связано с действием следующих причин:
А. Избыток соматотропина.
Б. Инфекционные заболевания.
В. Беременность.
Г. Период роста организма.
5. Сгруппируйте по соответствию буквы и цифры. А –
положительный азотистый баланс.
Б – отрицательный азотистый баланс.
1. Из организма выводится больше азота, чем
поступает с пищей.
2. Из организма выводится меньше азота, чем
поступает с пищей.
6. Сгруппируйте по соответствию буквы и цифры: А –
положительный азотистый баланс.
Б – отрицательный азотистый баланс.
1. Травмы.
2. Термические ожоги.
3. Избыток соматотропина.
4. Избыток тироксина.
7. Сгруппируйте по соответствию буквы и цифры,
отражающие (А) анаболический (усиление процессов
синтеза белка) и (Б) катаболический эффекты
(усиление процессов распада белка) под действием
следующих гормонов.
1. Тироксин.
2. Половые гормоны.
3. Соматотропин.
4. Инсулин.
5. Глюкокортикоиды.
8. Выберите правильные утверждения. К основным
нарушениям белкового состава крови относят:
А. Гипопротеинемия.
Б. Гипергликемия.
В. Гиперпротеинемия.
Г. Аминоацидурия.
Д. Парапротеинемия.
Е. Диспротеинемия.
9. Выберите правильное утверждение. К основным
причинам нарушения – снижение процессов
синтеза белка, относят:
А. Несбалансированное по аминокислотному
составу питание.
Б. Голодание.
В. Нарушение усвоения пищевых белков.
Г. Ожоги (плазморрагия).
10. Выберите правильное утверждение. К
основным причинам нарушения – снижение
процессов синтеза белка относят:
А. Дыхательная недостаточность.
Б. Воспалительные процессы (увеличение
проницаемости стенок капилляров).
В. Авитаминозы.
Г. Опухоли.
Д. Заболевания печени.
11. Выберите правильное утверждение. К
основным причинам, вызывающим избыточную
потерю белка организмом, относят:
А. Голодание.
Б. Ожоги (плазморрагия).
В. Воспалительные процессы, осложненные
свищами.
Г. Заболевания печени.
Д. Заболевания почек.
12. Выберите правильное утверждение. К
основным причинам, вызывающим потерю белка
организмом, относят:
А. Кровопотерю.
Б. Нефрозы.
В. Авитаминозы.
Г. Опухоли.
Д. Ожоги.
13. Выберите правильное утверждение. К
основным механизмам возникновения
гипопротеинемии относят:
А. Усиление процессов синтеза белка.
Б. Уменьшение поступления белка.
В. Избыточное поступление белка.
Г. Нарушение синтеза белка.
Д. Избыточную потерю белка организмом.
14. Выберите правильное утверждение.
Гипопротеинемия может приводить к следующим
патологическим процессам:
80