Hatano Y., Katsumura Y., Mozumder A. (Eds.) Charged Particle and Photon Interactions with Matter - Recent Advances, Applications, and Interfaces
Подождите немного. Документ загружается.


290 Charged Particle and Photon Interactions with Matter
12.1 introduCtion
Among all the radiation–chemical reactions that have been studied in solution, the most impor-
tant is the decomposition of pure water, the “life solvent” itself. At the beginning of the last cen-
tury, fruitful observations were made on the radiation chemistry of water (Giesel, 1900; Curie and
Debierne, 1901; Kernbaum, 1909; Duane and Scheuer, 1913). An important breakthrough is due to
Fricke’s studies on x-ray effects that appeared to be very similar to those of γ-rays (Fricke, 1934).
Since that demonstration, a wealth of data has been obtained to understand the mechanisms of water
decomposition under irradiation, particularly after the advent of nuclear energy and the extensive
research program around the Manhattan Project (Allen, 1989). One of the singular aspects of radia-
tion chemistry is energy deposition in the media, characterized by linear energy transfer (LET). In
contrast to UV–visible and IR light absorption that is homogeneous, the energy of ionizing radiation
is nonhomogeneously deposited in solutions. The direct visualization of α-particles branch tracks
inthe Wilson cloud chamber demonstrated the nonhomogeneity of the energy deposition. The mac-
roscopic visualization of such an effect can also be performed with the electron beam of an accel-
erator focused on a transparent block of polymers (Figure 12.1) (Gross, 1957, 1958). This important
aspect in radiation physics and chemistry led to the development of kinetics of nonhomogeneous
processes at the beginning of the last century (Freeman, 1987). When an ionizing radiation passes
through a solution, it loses energy and generates excited or ionized molecules. The ejected electrons,
called secondary electrons, possess enough kinetic energy to further ionize and excite the molecules
and to break and build chemical bonds. The initial event, energy deposition, classied as radiation
Figure 12.1 This Captured Lightning
®
sculpture is created by irradiating a block of acrylic (Plexiglas)
by a beam of electrons from a 3 MV particle accelerator. The rst Lichtenberg gures were actually two-
dimensional patterns formed in dust on the surface of charged insulating plates in the laboratory of their dis-
coverer, German physicist Georg Christoph Lichtenberg (1742–1799). Pioneering research on the detailed
behavior of charge storage and movement within dielectrics was performed by Bernhard Gross in the early
1950s. Gross conrmed that internal Lichtenberg gures could be created within a variety of polymers and
glasses by injecting them with high energy electrons using a linear accelerator (LINAC).
Time-Resolved Study on Nonhomogeneous Chemistry Induced in Polar Solvents 291
physics, is followed by a sequence of events such as ionization and excitation processes that lead to
the nal products of solvent decomposition. The yields of the nal products depend on the energy
distribution in the solvent. In solvents, the primary free radicals formed are assumed to be distrib-
uted in small volumes, called spurs, favoring radical–radical reactions according to the mechanism
rst suggested by Weiss (1944) and Allen (1948). For low LET, it is assumed that the spurs are
separated by large distances relative to their diameter that is around a few nanometers, and that in
each spur a few tens of eV energy is deposited. If we consider that the average energy in a spur is
around 40eV, the average separation of spurs along the track is 80/LET. For low LET (0.2eV nm
–1
)
the spurs are hundreds of nanometers apart (Magee and Chatterjee, 1987). To elucidate this special
feature of energy deposition, one of the decisive points was the observation that the yield of the nal
products depends on the LET, that is, on the energy distribution in the solvent. During World War II,
the idea of the spur and track structure was successfully supported by nding smaller radical yields
(for
e
s
−
, H
•
, and OH
•
) and larger molecular yields (H
2
and H
2
O
2
) with high LET radiolysis.
To follow the fast radiation-induced reactions in solvents, researchers have developed tools to
deliver ionizing radiations for their scientic studies. The advent of electron accelerators generated
the rst pulse-radiolysis experiments in the 1960s. Then, the knowledge on water decomposition
allowed the use of water radiolysis to trigger other reactions in a controlled manner. So, water and
more generally solvent radiolysis provide a very versatile method of generating radicals in a quanti-
tative way. The species issued from the solvent radiolysis can produce, in solution, different radicals,
redox states, or new chemical bonds through reactions with solutes or other solvent radicals. The
key point in these studies is the knowledge of the yields of the radical and molecular products at a
given time after energy deposition. The reactions with solutes at low concentration occur usually in
the homogeneous steps, that is, a hundred nanoseconds after energy deposition by ionizing radiation
with low LET such as γ-rays or accelerated electrons. The yields at the homogeneous step depend
on the initial distribution and chemical reactivity of the different radicals and molecules formed by
irradiation, that is revealed by the knowledge of the yield in the time window before the homoge-
neous step. Today, the yields at the homogeneous step are accurately known for the different radicals
and molecules issued from water irradiation, but their values in the subnanosecond time window
are
still debated.
To
elucidate the mechanism of solvent ionization and spur reactions, two approaches are pos-
sible. In the rst approach, the various radiation–chemical yields at different times can be esti-
mated by the use of solutes at high concentrations that selectively scavenge the radicals induced by
irradiation in the solvent, and by the analysis of the products. The second method consists of time-
resolved measurements to probe the media at different times after energy deposition and to follow
the radicals or the product of the radical scavenging using the spectrophotometric technique. In
his book published in 2004, Buxton (2004) presented an overview of the radiation chemistry of
water at room temperature and atmospheric pressure with low LET from the start of its systematic
study. To demonstrate the time evolution of the radical’s yields, he presented mostly the results
concerning the scavenging method. Since the publication of Buxton’s chapter, new results within
the short-time domain after energy deposition in the solvent have been reported thanks to the
development of new accelerators dedicated to pulse radiolysis and the amelioration of the detec-
tion systems.
In this chapter, we focus on the chemistry induced by irradiation before the homogeneous step
when the spurs are present in the solvent. The kinetics in water and also in some polar solvents,
ranging from a few hundred femtoseconds to a few nanoseconds, is discussed. After a general dis-
cussion of solvent decomposition under ionizing radiation, the recent time-resolved measurements
concerning the solvation dynamics of thermalized electron generated in water and other polar sol-
vents are discussed rst. New results are reported to discuss this very fast step that is usually studied
by femtosecond laser spectroscopy. Then, recent progress in the picoseconds pulse-radiolysis tech-
nique is presented and new data concerning the time-dependent yields of hydrated electron,
e
hyd
−
,
and
hydroxyl radical, OH
•
, are reviewed.
292 Charged Particle and Photon Interactions with Matter
12.2 solvent deComposition
A cascade of events is initiated by the absorption of energy by solvent from the incident ionizing
radiation. The rst step, shorter than 10
−16
s, corresponds to the formation of an excited-state elec-
tron and parent cation. For a given solvent denoted as RH, the ionizing radiation with low LET
induces RH
•
+
, RH
*
, and e
−
within this step (Figure 12.2) in small clusters called spurs that on aver-
age contain two or three ion pairs. The ejected electron generally has sufcient energy to ionize
or further excite solvent molecules. Mozumder (2002) reported that ionization potential in liquid
water dened as the minimum energy required to generate a quasi-free electron in the band gap has
a value of 11.7 ± 0.2 eV (see Figure 12.3). Nevertheless, low-energy processes, down to 6.5 eV,
also result in a small yield of
e
aq
−
via optical charge transfer or photoinduced electron transfer
RH
γ, X, e
–
RH*,
RH
+
, e
–
e
th
–
Electron attachment
ermalization
e
–
-molecule
Recombination
Recombination
Ion–molecule
Dimerization
Recombination
e
solv
–
Polymerization
R
–
H
2
, R
H
2
, R
2
(RH)
n
+
Polymerization
Dissociation
Deactivation
Solvation
RH
–
(RH)
n
–
R , H
RH
2
+
, R
R , H , e
th
–
• •
•
•
•
•
• •
Figure 12.2 General scheme for the chemical events occurring after energy deposition in RH solvent.
The main reactions in polar solvent are indicated with bold arrows.
Valence
band
Ground state of hydrated electron
Vacuum
Conduction band
2p
1s
3.2 eV,
Electron
stabilization
in cavity
6.5 eV
Photodissociation threshold
1.45 eV
1.7 eV
Hydration
energy
–10
–8
–6
Energy
–4
–2
0
Figure 12.3 Energy levels of liquid water and hydrated electron.
Time-Resolved Study on Nonhomogeneous Chemistry Induced in Polar Solvents 293
(Sander et al., 1993). Laser experiments performed by Bartels and Crowell (2000) with photon
energy up to 9.3eV support a concerted proton-coupled electron transfer mechanism, rst proposed
by Keszei and Jay-Gerin (1992). When the secondary electron loses the rest of its excess energy, the
so-called thermalized electron is formed. The excited states of the solvent molecule undergo disso-
ciation processes and the radical cation reacts quickly with another solvent molecule to form
RH
2
+
and R
•
. Solvation processes of electrons and recombination reactions between different species take
place in the spurs in parallel with diffusion of radicals out of spurs. This competition controls the
yields of the different products in the homogeneous step. The time of the end of spur reactions
depends
on the solvent and temperature.
In
the case of water, the current state of knowledge can be summarized by the reactions reported
in Figure 12.4. In less than 1 as (attosecond) after energy deposition, H
2
O
*
, H
2
O
•
+
, and e
−
are gener-
ated in the solvent. As the rate constant of the ion–molecule reaction is known to occur in the gas
phase with a rate of 8 × 10
12
dm
3
mol
−1
s
−1
, the positive radical ion H
2
O
•
+
is believed to undergo the
ion–molecule reaction in approximately 10
−14
s. In fact, the reaction between H
2
O
•
+
and H
2
O occurs
without any diffusion in the prethermal step. In the past, the observation of contribution of the water
cation, H
2
O
•
+
, in bulk water at 460nm was stated (Gauduel etal., 1990; Gauduel, 1995). But we
know that the very fast proton transfer reaction leads into H
3
O
+
and OH
•
radical in less than 10fs.
Therefore, perhaps an ultrashort pulse of a few attoseconds or femtoseconds is needed to observe
the H
2
O
+
cation in bulk water. Up to now, such an experiment has not yet been carried out. The
ejected electron remains in interaction with OH
•
and H
3
O
+
before its full solvation. Electron solva-
tion dynamics has been studied by several groups and will be discussed in Section 12.3. The elec-
tronically excited states H
2
O
*
are known to dissociate, as in the vapor phase, mainly into OH
•
and H
•
radicals, but the contribution of this channel is less important than the ionization of water molecule.
Another way of water dissociation is electron attachment occurring with electrons having an energy
of 5–15eV and giving H
−
and OH
•
. The hot H
−
ion can produce an electron by detachment, resulting
in dissociative electron attachment of a water molecule. But in that case, H
•
and OH
•
are generated
separately at a greater distance compared to a direct dissociation process. The H
−
ion can also react
Dissociation
H +
H
2
H
2
H
2
O
2
Homogeneous kinetics Nonhomogeneous kinetics
H
2
O
S
Stable product of
S reactions
10
–4
10
–7
10
–10
10
–13
10
–16
Time (s)
S
S
S
S
O
2
OH
H
2
O*,
H
2
O
H
3
O
+
H
3
O
+
OH
–
H
2
O
2
Ion–molecule
Solvation
H
2
O
H
2
O
e
–
e
aq
–
e
aq
–
H
2
O
+
,
OH
OH
H
Recombination
Dimerization
Dimerization
+
Figure 12.4 Mechanism of water decomposition under ionizing radiation.
294 Charged Particle and Photon Interactions with Matter
directly with H
2
O, producing unscavengable H
2
(Goulet and Jay-Gerin, 1989; LaVerne and Pimblott,
2000; Mozumder, 2005).
The H
3
O
+
, OH
•
, H
•
, and
e
hyd
−
species formed inside the spurs a few picoseconds after energy
deposition begin to diffuse randomly; so, a fraction of them encounter one another and react to
form molecular and secondary radical products, while the remainder escape into the bulk liquid
and become homogeneously distributed with respect to reaction with solutes acting as radical scav-
engers. In water, almost 100ns are needed to complete the nonhomogeneous kinetics and obtain a
homogeneous distribution of the different radicals and ions in the bulk solvent.
12.3 solvation proCesses oF eleCtrons in polar solvents
Two centuries ago, the solvated electron was rst observed in liquid ammonia (Thomas etal., 2008),
and a century later the idea of electrons trapped in cavities created by solvent molecules and behav-
ing like an anion was introduced (Kraus, 1908). It was only in 1962 that pulse-radiolysis experiments
gave evidence of the existence of the solvated electron by the observation of its optical absorption
spectrum (Hart and Boag, 1962). This discovery triggered many experimental and theoretical stud-
ies, as the solvated electron is a major transient species in condensed phase radiation chemistry.
However, the solvated electron continues to raise several fundamental questions, in particular on its
precursors, the nature of the “cavity” structure, and dynamics of the electron–solvent interactions.
In a simplied picture, the solvated electron structure resembles that of the hydrogen atom with a
ground
state of s-type character and three nondegenerate p-type excited states.
The
solvated electrons are generally formed by photoemission with ionizing radiation (radiolysis
or photolysis), and the early steps in electron solvation are depicted in a simplied manner in Figure
12.5. Knowledge of the timescale involved in solvated electron formation is crucial to understand
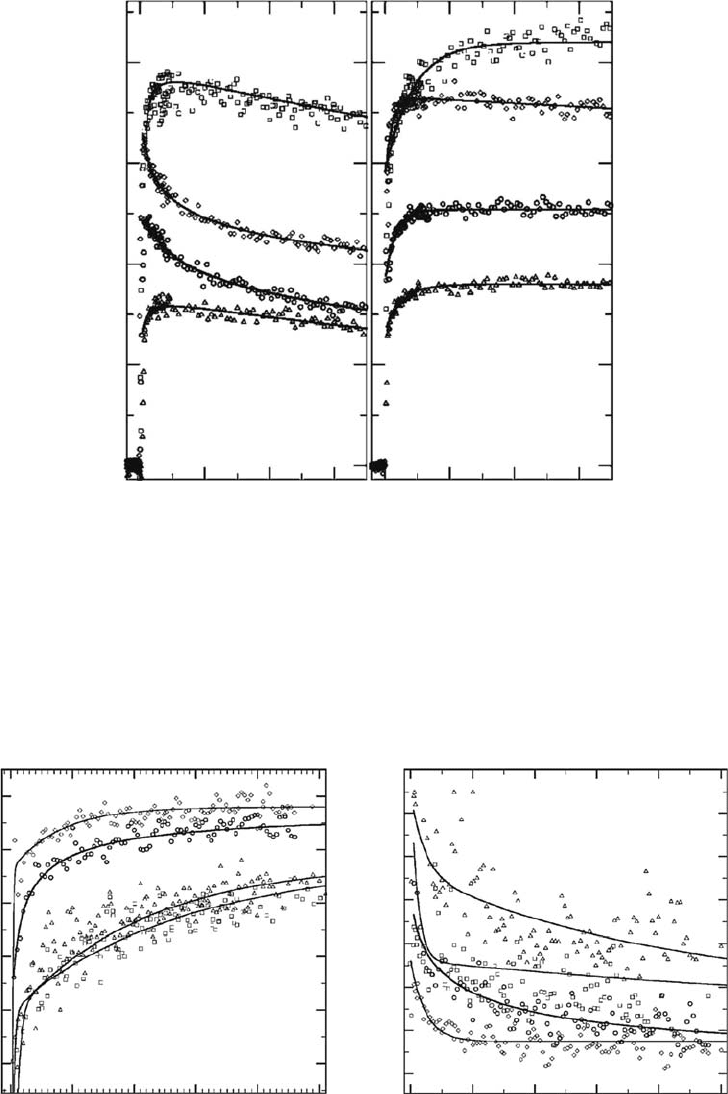
electron solvation and reactivity. Indeed, among the electron scavengers, some such as selenate
or molybdate anions are known to scavenge precursors to the solvated electron efciently and the
solvated electron poorly (Figure 12.6) (Pommeret etal., 2001). Others react with both precursors
and solvated electrons with similar efciencies, for instance
NO
3
−
or Cd
2+
(Kee etal., 2001), while
the remaining electron scavengers react poorly with the precursors but efciently with the solvated
electron
(Pastina etal., 1999).
Studies
on electron solvation began in the 1970s, rst in liquid alcohols at low temperatures by
pulse-radiolysis measurements (Baxendale and Wardman, 1971; Chase and Hunt, 1975), because by
decreasing the temperature, the molecular movements are slowed down and the electron dynamics
last several nanoseconds. Then, the emergence of ultrashort laser pulses made the investigation of
electron formation and early reactivity at room temperature possible (Nikogosyan etal., 1983). The
pioneering pump-probe laser experiments were performed in 1987 by Migus etal. (1987) in water.
They showed that the hydration process lasts less than 1ps and they gave evidence of precursors
(a) (b) (c) (d)
e
–
e
–
e
–
+
×
•
•
•
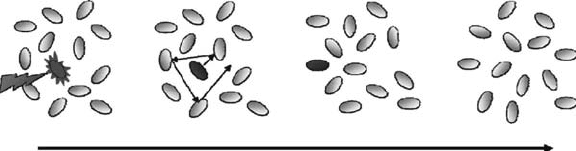
Figure 12.5 Formation of solvated electron by photoemission with ionizing radiation: (a) ionization, ejec-
tion of an electron from a molecule; (b) thermalization, loss of the excess kinetic energy of the electron
by collisions with solvent molecules; (c) localization, trapping of the electron in a solvent cavity; (d) solva-
tion, relaxation, and reorganization of the solvent molecules around the electron to reach the equilibrium
conguration.
Time-Resolved Study on Nonhomogeneous Chemistry Induced in Polar Solvents 295
of the hydrated electron absorbing in the infrared spectral domain (Figure 12.7). Since then, many
light-absorbing species or states were identied using decomposition analysis of transient absorption
spectra: “hot” s-like states, excited p-like states, “dry”/ “wet” electrons, “weakly bound” electrons,
etc. For the kinetic/spectral analyses, two main solvation models have been usually employed: step-
wise mechanisms (Hirata and Mataga, 1990; Long etal., 1990; Sander etal., 1992; Shi etal., 1995;
Reuther etal., 1996; Assel etal., 1999; Holpar etal., 1999; Kambhampati etal., 2002; Scheidt and
Laenen, 2003; Thaller etal., 2006) and continuous blueshift model (Hertwig etal., 1999; Kloepfer
etal., 2000; Madsen etal., 2000). The rst mechanisms consider a small number of species/states
with well-dened, time-independent spectra (Figure 12.8), while the second model consists of only
one species with a spectrum shifting to shorter wavelength but keeping its shape. In some instances,
both kinds of models were combined to interpret the data (Pépin etal., 1994, 1997; Turi etal., 1997;
Goulet
etal., 1999). However, the assumptions made in such models are not obvious.
Recently,
the formation of solvated electrons was investigated by photoionization in four polyal-
cohols, ethane-1,2-diol (Soroushian etal., 2006), propane-1,2-diol, propane-1,3-diol (Bonin etal.,
2007), and propane-1,2,3-triol (Bonin etal., 2008). In the diols, just after the pump pulse, the
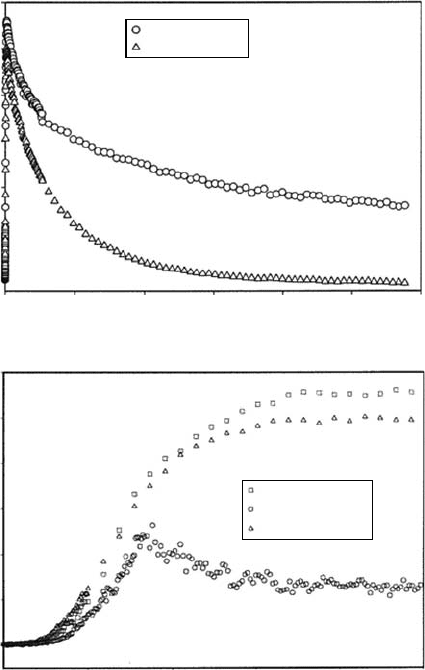
0
0.0
0.2
Transient absorbance
0.4
0.6
0.8
1.0
1.2
50 100 150
Pure water
[H
3
O
+
]=1.5 M
Pure water
[SeO
4
2–
]=1.0 M
[H
3
O
+
]=1.5 M
2000
0.0
0.2
0.4
0.6
Transient absorbance
0.8
1.0
1.2
400 600
Time (fs)(b)
(a)
800 1000
Time (ps)
200 250 300
Figure 12.6 Reactivity of the hydrated electron and its precursor with the hydronium cation H
3
O
+
(a) and
the selenate anion
SeO
4
2−
(b); transient kinetics measured at 800nm after the photoexcitation of aqueous solu-
tions
at 266
nm.
(Reprinted from Pommeret, S. etal., J. Phys. Chem. A, 105, 11400, 2001. With permission.)
296 Charged Particle and Photon Interactions with Matter
500
5
10
ΔA (10
–3
)
15
700 900
λ (nm)
1100 1300
2
1
0.7
0 ps
0.08
0.4
0.2
Figure 12.7 Absorption spectra of the electron at different delays after photoionization of liquid water at
21°C by two-photon excitation with pulses of 100fs duration at 310nm. (Reprinted from Migus, A. etal., Phys.
Rev. Lett.,
58, 1559, 1987. With permission.)
–6
(a) (b)
–3
–2
–1
0
E (eV)
|e*
free
c
|e*
free
G
|S
hot
G
|S
eq
C
75
60
45
30
15
0
20001000500
125 fs
0.6 ps
G
500
0
10
20
0
10
20
0
10
20
2.5 2.0 1.5
Energy (eV)
σ (10
3
M
–1
cm
–1
)
1.0 0.5
γ(°)
1000
Wavelength (nm)
20001500
Continuum
3.5 ps
7.9 ps
|p΄
G
|p˝
C
|p˝
G
|p˝
G
|S
hot
G
|S
eq
G
CH
3
OH
Figure 12.8 (a) Level diagram for the relaxation channel G of the kinetic model relevant for the photoion-
ization experiment of methanol with two-photon energy of 9.1eV. Terminating states of the probe absorption
are omitted. Vertical double arrow marks the excitation transition. (b) Spectral signatures of four intermedi-
ates in the photoionization process and of the nal solvated electron as deduced from the signal transients
(isotropic component) with the help of the kinetic model G. All spectra are normalized to the molar extinction
coefcient of solvated electrons in methanol; experimental points, calculated curves. (Reprinted from Thaller,
A.
etal., J. Chem. Phys., 124, 024515, 2006. With permission.)
Time-Resolved Study on Nonhomogeneous Chemistry Induced in Polar Solvents 297
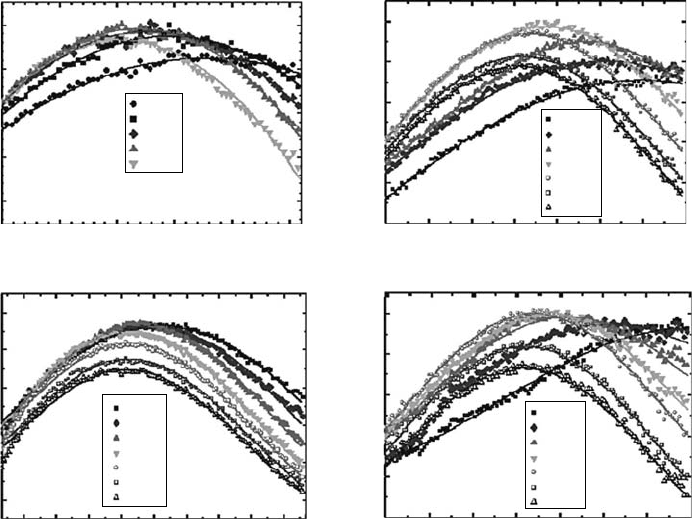
electron absorbs in the visible and near-infrared (NIR) domain and then, for the rst few picosec-
onds or tens of picoseconds, the red part of the spectrum goes down while the blue part increases
slightly, inducing a blueshift of the spectra. In contrast, in the triol, the electron absorption spec-
trum is situated early on in the visible domain and evolves only on the red side (Figure 12.9). The
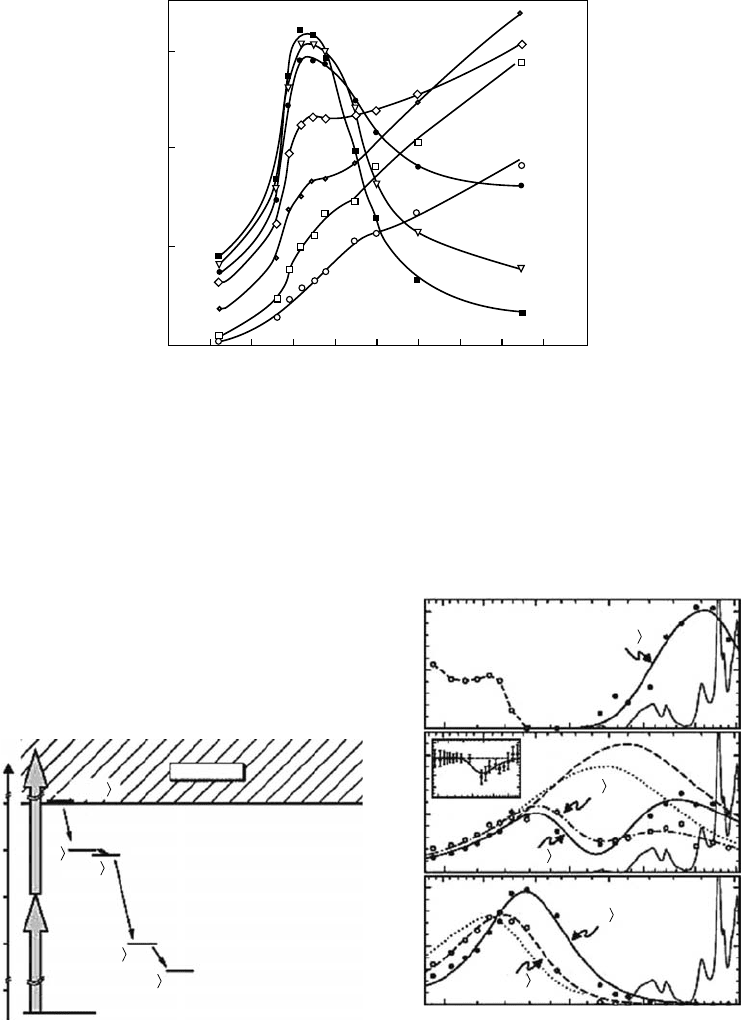
kinetics signals observed for the four polyols at 700nm and at the wavelength of the maximum
of the fully solvated electron band focus on the change in the electron spectra on the red side and
highlight distinct evolutions for the four polyols (Figure 12.10). More closely, the time evolution of
the peak position and the width of the electron spectra show that the solvation dynamics is different
in the two propanediols and is much faster in the propanetriol despite its higher viscosity (Figure
12.11). Those results clearly show that the solvation dynamics cannot be explained by the viscosity
alone but also depends on the molecular structure of the solvent, in particular on the OH groups. To
extract the characteristic times of the solvation process, it is essential to analyze the full set of tran-
sient absorption spectra. For that, Bayesian data analysis is a powerful tool (Sivia, 1996; D’Agostini
2003). This probabilistic method, mostly used in environmental modeling (Brun etal., 2001) and
biokinetics (D’Avignon etal., 1998, 1999; Holtzer etal., 2001), provides a consistent framework
for optimization and identication of independent parameters, determination of uncertainties, and
model comparison. So, the results of the Bayesian data analyses performed for the four polyols and
comparing various models are in favor of a modied continuous “blueshift” model considering
a unique species with an absorption band changing continuously not only in position but also in
shape. These analyses are corroborated by the analogy between the time evolution of the electron
absorption band at a given temperature and the change in the spectrum of the solvated electron by
decreasing the temperature. A similar type of dynamics was observed in liquid water by Lian and
0.14
0.27
0.24
0.21
0.18
0.15
0.12
440 480 520 560 600 640 680 720
0.12
0.10
0.08
0.06
0.16
0.14
0.12
0.10
0.08
0.06
450
450 500 550
Wavelength (nm) Wavelength (nm)
440
0.10
0.12
0.14
0.16
0.18
0.20
480 520 560 600 640 680 720
Wavelength (nm)
12ED
123PT
13PD
12PD
600 650 700
480 510 540 570
Wavelength (nm)(b)
(a)
(d)
(c)
1 ps
3 ps
10 ps
50 ps
100 ps
250 ps
440 ps
1 ps
3 ps
15 ps
50 ps
Absorbance
Absorbance
Absorbance
Absorbance
100 ps
250 ps
450 ps
1 ps
5 ps
10 ps
50 ps
0.5 ps
2 ps
5 ps
15 ps
40 ps
100 ps
230 ps
450 ps
600 630 660 690
Figure 12.9 Time evolution of the absorption spectrum obtained in four polyols upon two-photon ioniza-
tion of the solvent at 263nm: (a) 12ED, ethane-1,2-diol; (b) 123PT, propane-1,2,3-triol; (c) 12PD, propane-
1,2-diol; (d) 13PD, propane-1,3-diol. (Reprinted from Lampre, I. etal., J. Mol. Liq., 141, 124, 2008a. With
permission.)
298 Charged Particle and Photon Interactions with Matter
0.20
0.15
0.10
0.05
0.00
0 10 20 30 0 10
Time (ps)Time (ps)
λ=700 nm
Absorbance
λ=λ
max
of e
s
–
20 30
Figure 12.10 Transient absorption signals recorded at 700nm and at the wavelength of the solvated elec-
tron absorption band maximum for the four studied polyols (Figure 12.9): ◇ 123PT, ○ 12ED, △ 13PD, and
□12PD. (Reprinted from Lampre, I. etal., Radiat. Phys. Chem., 77, 1183, 2008b. With permission.)
2.3
2.2
2.1
2
1.9
1.8
2.6
(b)(a)
2.4
2.2
2.0
1.8
1.6
1.4
1.2
0 10 20
Time (ps)
Ω (eV)
E
max
(eV)
30 40 500 10 20
Time (ps)
30 40 50
Figure 12.11 Time evolution of the peak position E
max
(a) and the width (b) of the absorption band of the
photogenerated excess electron in the four studied polyols (Figure 12.9): ◇ 123PT, ○ 12ED, △ 13PD, and □
12PD.
(Reprinted from Bonin, J. etal., J. Phys. Chem. A, 112, 1880, 2008. With permission.)
Time-Resolved Study on Nonhomogeneous Chemistry Induced in Polar Solvents 299
coworkers (Lian etal., 2005) with two regimes: rst, a rapid shift of the spectrum with a change
in shape on the subpicosecond timescale, then, a slower narrowing of the spectrum on the red side
while the position of the maximum remains xed. Such behavior looks like vibrational relaxation in
photoexcited molecules and the presence of a short lived peak at 1.2μm, overtone of the OH stretch
mode,
indicates a strong vibronic coupling between the electron and the water solvent.
Those
results strongly suggest that in femtosecond solvent photoionization (or electron photo-
detachment), electrons are very quickly trapped and only localized electrons in s-like states are
observed. Indeed, while probing the electron delocalization in liquid water and ice at attosecond
timescales, Nordlund etal. found that the trapping of the electron at a broken H-bond provides a
timescale (20fs) long enough for librational response of the water molecules and early-time dynam-
ics leading to the hydrated electron (Nordlund etal., 2007). Moreover, time-resolved resonance
Raman experiments performed in water gave evidence of enhanced Raman signals for the electron
precursor (Mizuno etal. 2005). Since intensity enhancement is due to s-p transition, the precursor is
assumed to be a nonequilibrium s-state electron. It is worth noticing that resonance Raman experi-
ments have provided valuable data on the structure of the solvated electron in water (Tauber and
Mathies, 2002, 2003; Mizuno and Tahara, 2003) and primary alcohols (Tauber etal., 2004), reveal-
ing strong vibronic couplings between the solvated electron and the normal modes of the solvent.
In the case of alcohols, couplings to the methyl/methylene deformations indicate that the electronic
wave function of the electron extend on the alkyl group of the alcohols. Those experimental results
agreed with the theory developed by Abramczyk and coworkers in which the coupling between the
excess electron and the solvent vibrational modes plays a signicant role in the electron dynamics
and absorption spectra (Abramczyk, 1991; Abramczyk and Kroh, 1992, 1994). The other concep-
tual picture, suggesting that the solvated electron is an unusual kind of solvent multimer anions
in which the excess electron density occupies voids and cavities between the molecules and fron-
tier p-orbitals in the heteroatoms in the solvating groups, can also account for these experimental
observations (Symons, 1976; Shkrob, 2006). Ultrafast relaxation dynamics of the solvated electron
in liquid ammonia solutions were also investigated by femtosecond NIR pump-probe absorption
spectroscopy (Lindner etal., 2006). Immediately, after photoexcitation, the absorption spectrum of
the electron is substantially redshifted with respect to its stationary spectrum, and a subsequent blue
shift occurs on a timescale of 150fs. The data are understood in terms of a temperature-jump model
and ground-state “cooling.” Spectral ngerprints of the excited p-state were neither unambiguously
observed nor required to explain the observed dynamical response indicating that its lifetime is very
short
(<100
fs).
If p-like states do not seem detectable upon photoionization or electron photodetachment, such
states may be generated by excitation of solvated electrons (Walhout etal., 1995; Assel etal., 1998,
1999; Silva etal., 1998a,b; Kambhampati etal., 2002; Thaller etal., 2006) and large water anions
(Bragg etal., 2004; Paik etal., 2004; Lee etal., 2008; Ehrler and Neumark, 2009). In bulk solvents,
two main schemes were proposed for the relaxation of p-like states: (1) slow adiabatic internal con-
version (IC) and (2) fast nonadiabatic IC, leading to a “hot” s-like state that relaxes subsequently.
In water, according to the rst scheme, the p-state lifetime is determined to be a few hundreds of
femtoseconds (Assel etal., 1998; Silva etal., 1998b; Yokoyama etal., 1998) after a fast excited-state
solvation dynamics, while with the second scenario, the IC time is around 50fs and the hundreds
femtosecond component is attributed to the initial relaxation of the “hot” s-like state (Pshenichnikov
etal., 2004). The second scenario is supported by photon echo and resonant transient grating mea-
surements (Emde etal., 1998) as well as time-resolved photoelectron experiments on anion clusters.
Indeed, cluster experiments indicate p → s IC time depending on cluster size n (Figure 12.12).
Extrapolation of these values to n → ∞ yields IC lifetimes of 50 and 157 fs for water and methanol,
respectively (Ehrler and Neumark, 2009). Paik and coworkers reported bi-exponential ground-state
relaxation, after IC (Paik etal., 2004). They found that the solvation time (300fs) was similar to that
of bulk water, indicating the dominant role of the local water structure in the dynamics of hydration;
but in contrast, the relaxation in other nuclear coordinates was on a much longer timescale (2–10ps)