Schlick T. Molecular Modeling and Simulation: An Interdisciplinary Guide
Подождите немного. Документ загружается.

420 12. Monte Carlo Techniques
can be developed, as in the “end-transfer CB-MC” for chromatin, where one end
of the polymer is grown at the other end. Dramatic efficiency can be achieved –
quadratic vs. exponential scaling – in such applications [63].
A comparison of several biased MC schemes for small molecule design [224]
demonstrated the excellent performance of such methods compared to genetic
algorithms.
12.6.3 Hybrid MC
Exploiting Strengths of MC and MD
To enhance the efficiency of MC simulations, a simple idea emerged that attempts
to combine the favorable properties of molecular dynamics (MD) simulations —
sampling phase space in a directed manner guided by the shape of the energy
gradient — with that of MC — sampling phase space more globally. Ideally,
following conformation space by MD would generate a correct Boltzmann dis-
tribution of states, but the relatively short simulation lengths that are possible (see
MD chapters) imply local rather than global sampling.
This MC/MD combination [344, 347, 688] — moving some particles by MC
rules and others by MD, for example by combining global updates in position
space via MD with reasonable acceptance criteria by MC — can be more effective
for solvated biomolecular systems than either MC or MD alone. Such hybrid MC
methods have been very effective for small systems [575].
Overall Idea
For example, the first step of a hybrid MC method can use a molecular dynamics
framework to specify the system’s candidate move: X[iΔt] −→ X[(i +1)Δt]
where Δt is the timestep. The MD algorithm must be time reversible and vol-
ume preserving so as to ensure detailed balance. The commonly used symplectic
Verlet method satisfies this requirement (see Chapters 13 and 14). Since the re-
cursive MD recipe relies on the velocities in addition to positions, the required
velocity vectors V = M
−1
P (P here designates momentum, not to be con-
fused with the symbol used earlier for probability) are generated from a Gaussian
distribution so as to obtain the target kinetic energy at the temperature T as-
suming energy equipartition. That is, the velocity components are drawn from a
Gaussian distribution proportional to the Boltzmann factor applied to the kinetic
energy:
ρ(P ) ∝ exp [−βE
k
(P )] . (12.46)
Following this MD-guided step, the second step of the hybrid MC method
applies the standard Metropolis acceptance criterion where the energy in the
Boltzmann factor is the Hamiltonian (potential plus kinetic energy). Namely,
the Metropolis criterion is applied to accept the new candidate
˜
X
i+1
with
probability

12.6. Monte Carlo Applications to Molecular Systems 421
p
acc, HMC,ρ
=min
1, exp
−β
H(
˜
X
i+1
,
˜
P
i+1
) − H(X
i
,P
i
)
=min
1,
exp [−βE
p
(
˜
X
i+1
)] exp [−βE
k
(
˜
P
i+1
)]
exp [−βE
p
(X
i
)] exp [−βE
k
(P
i
)]
=min
1,
ρ
NVT
(
˜
X
i+1
)exp[−βE
k
(
˜
P
i+1
)]
ρ
NVT
(X
i
)exp[−βE
k
(P
i
)]
. (12.47)
Note that generalized ensembles can be emulated by HMC methods by ap-
plying weighting factors to the Metropolis-generated configurations. That is,
to sample from a general ensemble with μ(x) rather than ρ(x), we adjust the
acceptance criteria of eq. (12.47)tobe:
p
acc, HMC,μ
=min
1,
μ(
˜
X
i+1
)exp[−βE
k
(
˜
P
i+1
)]
μ(X
i
)exp[−βE
k
(P
i
)]
. (12.48)
The weighting must be accomplished to maintain detailed balance.
HMC methods have been quite successful for biomolecular sampling, and
many extensions to general ensembles and hybrid methods have been described.
See [15], for example, for a comparison of several HMC methods with various de-
tailed balance conditions, and [596] for a shadow HMC method which improves
sampling while adding modest computational cost.
12.6.4 Parallel Tempering and Other MC Variants
One idea that has gained popularity involves temperature jumps to accelerate
sampling. This idea of replica exchange is similar to simulated annealing but
here multiple simulations of non-interacting systems are involved [508, 1237].
Specifically, replicas (multiple copies) of the system that do not interact with one
another are simulated at different temperatures, and systems from different en-
sembles are periodically exchanged with a transition probability that maintains
each temperature’s equilibrium ensemble distribution:
p
acc, MC−PT
=min[1, exp ((β
j
− β
i
)(E
j
− E
i
)) ] (12.49)
where β
i
=1/(k
B
T
i
).
REM, however, requires many parallel processors and a careful protocol of
ensemble exchange and parameter selection to obtain rapid sampling and conver-
gence. These issues have been addressed mostly in the dynamics analog of REM
termed REMD. See a separate discussion of these issues in the MD chapters.
An energy-restricted multiple random walk MC protocol with similar advan-
tages to REM tempering in escaping from local barriers was introduced by Wang
& Landau [1328]. The method performs multiple random walks in energy space,
each to sample a different range of energy; the resulting information is combined
422 12. Monte Carlo Techniques
to produce canonical averages for calculating thermodynamic quantities at any
temperature. Both this method and REM perform similarly for protein conforma-
tional sampling and are faster by two orders of magnitude when compared to a
canonical MC simulation at a low temperature. However, the Wang/Landau MC
method was found to be easier to implement on single-processor systems, while
parallel tempering is advantageous for multi-processor machines [623].
Additional MC variants include Smart MC [1162], stochastic dynamics, um-
brella sampling, various methods that incorporate MC elements to enhance
barrier crossing events (e.g., the transition path sampling method of Chandler
and coworkers [147]), and potential-modification approaches such as smoothing
and potential biasing techniques. See separate section for enhanced sampling and
recent reviews [1117,1117].
Given recent successes of MC methods for biomolecules [347,781,1457], some
advocate that recent improvements in MC methodology and increased computer
memory and speed lend support for the increased application of MC algorithms
for folding small biomolecules. Indeed, canonical, multi-canonical, biased MC
protocols that incorporate experimental information (e.g., knowledge-based di-
hedral angle distributions, and hybrids involving global optimization techniques
and MD) can significantly enhance the sampling of low energy configurations and
reveal folding ensembles of small proteins.
General and flexible MC modules have been built into standard programs like
CHARMM [575], with automatic optimization of step sizes and efficient combi-
nations with minimization or MD modules. These optimized MC methods were
found to outperform standard Langevin dynamics simulations in reaching folded
states of small proteins.
In general, MC methods can become inefficient for large systems but can
certainly be effective for coarse grained methods (e.g., chromatin folding [64,
1240]) and as vital components of other methods (e.g., transition path sampling
[147]). With increased usage and interest in coarse-grained models for larger
biomolecular complexes, further development of MC methods, including the
various extensions and hybrids described above, is warranted for biomolecular
applications as a whole.
There has been continued discussion on the relative merits of MC and MD for
biomolecules (e.g., [247, 619]). The limits of MD for simulating slow molecu-
lar events are clearly recognized, as is the high computational cost, though only
MD can ultimately yield detailed dynamic information such as folding pathways
and rates of conformational changes. Today, many enhanced sampling tools are
available (discussed separately). Thus, a practitioner is well-advised to use a com-
bination of MC, MD, local and global minimization algorithms, and enhanced
sampling approaches as appropriate, for the problems at hand.

12.6. Monte Carlo Applications to Molecular Systems 423
13
Molecular Dynamics: Basics
Chapter 13 Notation
S
YMBOL DEFINITION
Matrices
M mass matrix
P
pressure tensor (stress)
Vectors
g(X) constrained dynamics vector, components g
i
= r
2
jk
− r
jk
2
r
i
position vector for atom i
v
i
velocity vector component corresponding to atom i
F
force
F
acceleration (
¨
X), or M
−1
F (X(t))
F
i
force on particle i due to all particles
V ,
˙
V ,
¨
V
velocity =
˙
X (components {v
i
}), and its first and second
time derivatives
V
0
initial velocity
X,
˙
X,
¨
X
position (components {x
i
}), and its first and second
time derivatives
X
0
initial position
Λ
Lagrange multiplier vector for constrained dynamics
(components λ
i
)
∇E
energy gradient
Scalars & Functions
c speed of light
c
p
,c
t
coordinate and velocity scaling parameters (for
constant pressure and temperature simulations)
h
Planck’s constant
m
particle mass
T. Schlick, Molecular Modeling and Simulation: An Interdisciplinary Guide, 425
Interdisciplinary Applied Mathematics 21, DOI 10.1007/978-1-4419-6351-2
13,
c
Springer Science+Business Media, LLC 2010

426 13. Molecular Dynamics: Basics
Chapter 13 Notation Table (continued)
S
YMBOL DEFINITION
m
p
,m
t
masses for fictitious thermostat and barostat dynamic
variables
r
ij
, r
ij
interatomic distance and associated equilibrium value
v
cm
velocity of the center of mass
x
p
,x
t
dynamic variables for fictitious thermostat and barostat
E
k
kinetic energy
E
p
potential energy
H,
H
Hamiltonian and nearby Hamiltonian
N
number of atoms
N
F
number of degrees of freedom
P, P
0
pressure, target pressure
S
h
harmonic bond stretching constant
T, T
0
temperature, target temperature
β
isothermal compressibility
γ
t
thermostat coupling parameter
δ(t)
Lyapunov exponent
trajectory perturbation
ζ
p
,ζ
t
thermodynamic coefficients for constant pressure and
temperature simulations
λ
wavelength (1/λ is wave number)
ν, ω
characteristic frequencies
ν
l
volume variable
τ
thermostat coupling parameter
Δt, Δt
m
, Δτ timesteps
Time is defined so that motion looks simple.
John Archibald Wheeler, American theoretical physicist (1911–2008).
Molecular dynamics is clearly on the way to being a universal tool,
as if it were the differential calculus.
Sir John Royden Maddox [811], British science writer and Nature editor
(1925–2009).
13.1 Introduction: Statistical Mechanics by Numbers
13.1.1 Why Molecular Dynamics?
Molecular dynamics (MD) simulations represent the computer approach to sta-
tistical mechanics. As a counterpart to experiment, MD simulations are used to
estimate equilibrium and dynamic properties of complex systems that cannot be
calculated analytically. Representing the exciting interface between theory and
experiment, MD simulations occupy a venerable position at the crossroads of
mathematics, biology, chemistry, physics, and computer science.
13.1. Introduction: Statistical Mechanics by Numbers 427
The static view of a biomolecule, as obtained from X-ray crystallography for
example — while extremely valuable — is still insufficient for understanding
a wide range of biological activity. It only provides an average, frozen view
of a complex system. Certainly, molecules are live entities, with their constituent
atoms continuously interacting among themselves and with their environment.
Their dynamic motions can explain the wide range of thermally-accessible states
of a system and thereby connect sequence to structure and function. Thus,
by following the dynamics of a molecular system in space and time, we can
obtain a rich amount of information concerning structural and dynamic proper-
ties. Though considered obvious today, relating motion to function of proteins
through sampling rugged energy landscapes was innovative when first described
by Frauenfelder and Wolynes [422,1386].
Indeed, following the dynamics of molecular systems can provide valu-
able information concerning molecular geometries and energies; mean atomic
fluctuations; local fluctuations (like formation/breakage of hydrogen bonds,
water/solute/ion interaction patterns, or nucleic-acid backbone torsion motions);
rates of configurational changes (ring flips, nucleic-acid sugar repuckering, diffu-
sion), enzyme/substrate binding; free energies; and the nature of various types
of concerted motions. Ultimately perhaps, large-scale deformations of macro-
molecules such as protein folding might be simulated, as discussed in the first
chapter. This formidable aspect, however, is more likely to be an outgrowth of
hand-in-hand advances in both experiment and theory, not to speak of high-end
computing.
Though the MD approach remains popular because of its essential simplic-
ity and physical appeal (see below), it complements many other computational
tools for exploring molecular structures and properties, such as Monte Carlo sim-
ulations, Poisson-Boltzmann analyses, and energy minimization, as discussed in
preceding chapters, and Brownian dynamics and enhanced sampling methods, as
described in the next chapter. See examples in Table 13.1 and Figure 13.1. Each
technique is appropriate for a different class of problems.
13.1.2 Background
A solid grounding in classical statistical mechanics, thermodynamic ensembles,
time-correlation functions, and basic simulation protocols is essential for MD
practitioners. Such a background can be found, for example, in the books by
McQuarrie [853], Allen and Tildesley [22], and Frenkel and Smit [428]. Basic ele-
ments of simulation can be learned from the book of Ross [1067] and Bratley, Fox
and Schrage [165]. MD fundamentals, as well as advanced topics, are also avail-
able in texts by Allen and Tildesley [22], Frenkel and Smit [428] and Berenedsen
[122]. Basic introductions and useful examples in Gould and Tobochnik [474],
Rapaport [1038], Haile [494], and Field [398]. Some of these texts also describe
analysis tools for MD trajectories, a topic not considered here.
Good advanced texts for Hamiltonian dynamics and integration schemes are
those by Leimkuhler and Reich [731] and by Sanz-Serna and Calvo [1090]. Two

428 13. Molecular Dynamics: Basics
Table 13.1. Selected biomolecular sampling methods. Continuum solvation includes em-
pirical constructs, generalized Born models, stochastic dynamics, or Poisson Boltzmann
solutions, as discussed in Chapter 10. Targeted MD involves generating hypothetical paths
between two conformational endpoints [389]. See Figure 13.1 for illustrations.
Method Pros Cons CPU
• Molecular
Dynamics
(MD)
continuous motion, experimental bridge
between structures and macroscopic ki-
netic data
expensive; short
timespan
high
• Targeted
MD (TMD)
connection between two states; useful for
ruling out steric clashes and suggesting
high barriers
not necessarily physi-
cal
moderate
• Continuum
Solvation
mean-force potential approximates envi-
ronment and reduces model’s cost; useful
information on ionic atmosphere and
intermolecular associations
approximate moderate
• Brownian
Dynamics
(BD)
large-scale and long-time motion approximate hydrody-
namics; limited to sys-
tems with small inertia
moderate
• Monte Carlo
(MC)
large-scale sampling; useful statistics move definitions are
difficult; unphysical
paths
low
•
Minimization
valuable equilibria information; experi-
mental constraints can be incorporated
no dynamic informa-
tion
low
books written in the early days of biomolecular simulations are by McCammon
and Harvey [846] and by Brooks, Pettitt and Karplus [178]. And there are always
good reviews of MD from time to time (e.g., [4,636,658]).
Given these many sources for the theory and application of MD simulations,
the focus in this chapter and the next is on providing a broad introduction
into the computational difficulties of biomolecular MD simulations and some
long-time integration approaches to these important problems. The reader is
advised to complement these aspects with the more standard topics available
elsewhere.
13.1.3 Outline of MD Chapters
We begin by describing in Section 13.2 the roots of molecular dynamics in
Laplace’s 19th-century vision. We then describe two basic limitations of Newto-
nian (classical) mechanics: determinism, and failure to capture quantum effects
(or electronic motion). We elaborate on the latter by noting that the classical
physics approximation is especially poor at lower temperatures and/or higher fre-
quencies. Section 13.3 discusses general issues in molecular and biomolecular
dynamics simulations, such as setting up the system (initial conditions, solvent
and ions, equilibration), estimating temperature, and other simple aspects of
the simulation protocol (equilibration, nonbonded interaction handling). We also
13.2. Laplace’s Vision of Newtonian Mechanics 429
elaborate upon the inherent chaotic (but bounded) behavior of biomolecules first
mentioned in Section 13.2 by demonstrating sensitivity to initial conditions. We
motivate various methods for reducing the computational time of dynamic simu-
lations by emphasizing the large computational times required for biomolecules,
as well as extensive data-analysis and graphical requirements associated with the
voluminous data that are generated.
Given the large computational requirements, we introducein the same overview
section the concepts of accuracy, stability, symplecticness, and multiple-timesteps
(MTS) that are key to developing numerical integration techniques for biomolec-
ular dynamics simulations. In this context, we also mention constrained dynamics
and rigid-body approximations.
Though we refer to several of the methods in this introductory section (Section
13.3) that we only define later, a detailed understandingis not needed for grasping
the essentials that are communicated here. I recommend that students re-read
Section 13.3 after completing the two MD chapters.
Section 13.4 describes the symplectic Verlet/St¨ormer method and develops
its variants known as leapfrog, velocity Verlet, and position Verlet. This ma-
terial is followed by extension of Verlet to constrained dynamics formulations
(Section 13.5) and a reference to various MD ensembles (Section 13.6).
The next chapter introduces more advanced topics on various integration ap-
proaches for biomolecular dynamics; the material is suitable for students with
a good mathematical background. Specifically, we discuss symplectic integra-
tion methods, MTS (or force splitting) methods — via extrapolation and impulse
splitting techniques — and introduce the notion of resonance artifacts. Enhanc-
ing our understanding of resonance artifacts in recent years has been important
for developing large-timestep methods for MD. We continue to describe methods
for Langevin dynamics, Brownian dynamics, implicit integration, and enhanced
sampling.
13.2 Laplace’s Vision of Newtonian Mechanics
13.2.1 The Dream Becomes Reality
Besides “statistical mechanics by numbers”, I like to term MD as “Laplace’s
vision of Newtonian mechanics on supercomputers”.
Sir Isaac Newton (1642–1727) described in 1687 in his Principia masterpiece
(PhilosophiaeNaturalis Principia Mathematica) the grand synthesis of basic con-
cepts involving force and motion. This achievement was made possible by the
large pillars of empirical data laid earlier by scientists who studied the motion
of celestial and earthly bodies: Galileo Galilei (1564–1642), Nicolas Copernicus
(1473–1543), Tycho Brahe (1546–1601), and Johannes Kepler (1571–1630).
Newton’s second law of motion — that a body’s acceleration equals the net force
divided by its mass — is the foundation for molecular dynamics.
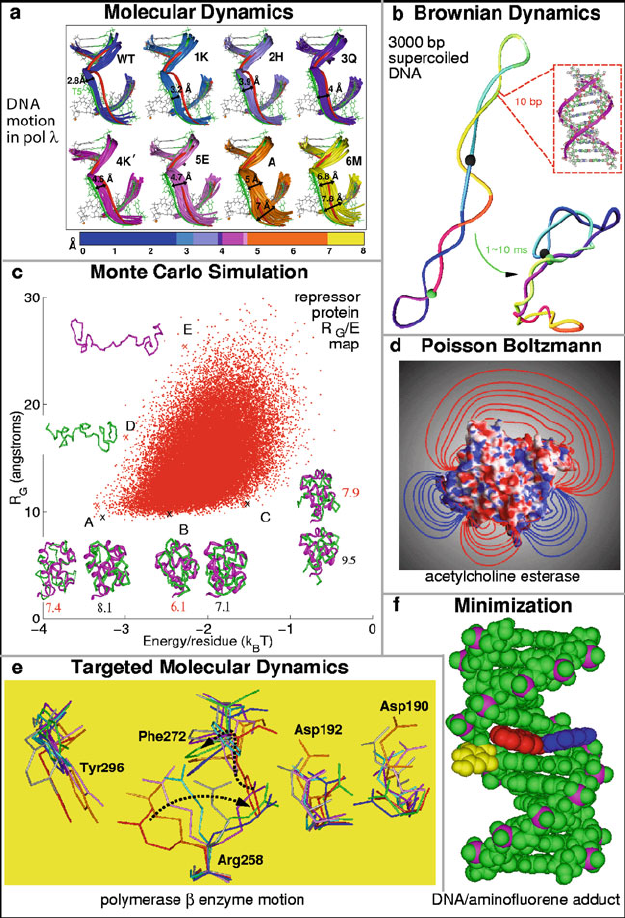
430 13. Molecular Dynamics: Basics
Figure 13.1. Illustrative images for selected biomolecular sampling methods. See caption
on the following page.