Schlick T. Molecular Modeling and Simulation: An Interdisciplinary Guide
Подождите немного. Документ загружается.

13.3. The Basics: An Overview 441
0 5 10 15 20
Time (ps)
Δτ = 1 fs
ε = ±
ε = ±
ε = ±
0 5 10 15 20
10
−8
10
−7
10
−6
10
−5
10
−4
10
−3
10
−1
10
−2
10
0
10
−8
10
−7
10
−6
10
−5
10
−4
10
−3
10
−1
10
−2
10
0
Time (ps)
RMS coordinate deviation (A)
RMS coordinate deviation
(A)
Δτ = 0.1 fs
ε = ± 10
−4
A
10
−4
A
10
−6
A
10
−8
A
10
−6
A
10
−8
A
ε = ±
ε = ±
Figure 13.3. The rapid divergence of dynamic trajectories for four water molecules
differing slightly in initial conditions.
This stabilization behavior realized in practice for MD simulations can also
be understood by the different mathematical limit involved in the strict notion of
chaos compared to that relevant for MD. Physicists define chaos rigorously for
finite times but in the limit of timesteps approaching zero; in MD applications, we
instead work with finite timesteps and simulation times approaching infinity.
Statistical View
For many other reasons than inherent chaos, including the approximate nature
of biomolecular simulations, MD trajectories are not intended to animate the
life of a biomolecule faithfully, as if by an unbiased video camera. Rather, our
experimental-by-nature computer ‘snapshots’ aim at gaining structural insights,
which can be tested, and predicting meaningful statistical properties of a complex
system in the ultimate goal of relating structure to function. The more reliable
quantities from MD simulations are thus space and time averaged. This is un-
like differential-equation applications in astrophysics, where precise trajectory
pathways are sought (e.g., planetary orbits). Often, more useful data are obtained
by averaging over, or calculating from, several trajectories rather than from one
long trajectory (e.g., [158, 286, 360, 364, 1428]). This has become very suitable
442 13. Molecular Dynamics: Basics
now with readily available parallel processor architectures and loosely connected
computer networks used for a common goal, as in the folding@home dis-
tributed computing initiative for simulating protein folding and related processes.
Launched in 2001, folding@home has become an international enterprise, with
clients like Sony PlayStations and numerous platforms. See also Skeel’s recent
analysis [1197] for a perturbation analysis on the effect of errors that aims to
show why molecular dynamics simulations “work”.
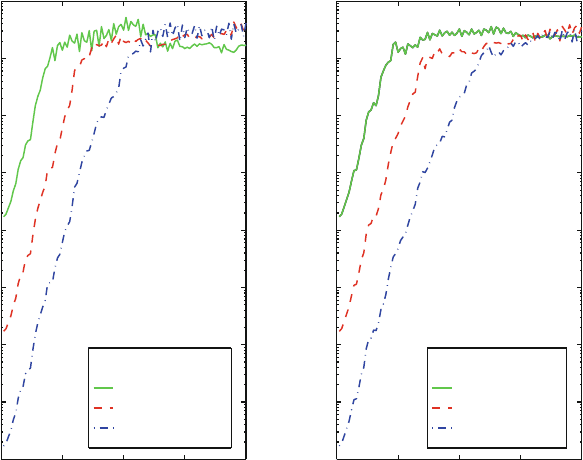
Let us consider the convergence of the end-to-end distance of a butane mol-
ecule, as first illustrated in [304]. Figure 13.4 shows the time evolution of the
end-to-end-distance of the butane molecule for different choices of timesteps in
the range of 0.02 to 2.0 fs. Clearly, different paths are realized even for this very
small system. The associated average end-to-end distance values as a function of
the timestep are the points corresponding to the dashed curve in Figure 13.5.
These data reveal convergence only for small timesteps, less than 0.2 fs. This
value is typically much smaller than standard timesteps used in biomolecular sim-
ulations (0.5 fs or greater). Still, larger values can be quite reasonable depending
on the specific questions being asked from a simulation.
13.3.5 Simulation Protocol
A careful simulation protocol is essential for MD simulations, to ensure not only
the equilibration phases but also proper enforcement of the boundary conditions
0 100 200 300 400 500 600 700 800 900 1000
7
7.5
8
8.5
9
9.5
10
10.5
11
11.5
Time (fs)
End−to−end distance (Angstrom)
0.02 fs
0.2 fs
1.0 fs
2.0 fs
Figure 13.4. Time evolution of the end-to-end distance of butane for different timesteps.
13.3. The Basics: An Overview 443
(when, for example, a biomolecule is placed in a box of water molecules), po-
sitioning of solvent and salt molecules, or computation of the nonbonded terms
and associated pairlist arrays. Also important is monitoring the kinetic tempera-
ture and the energy fluctuations, for the detection of systematic drifts or abrupt
changes, both of which may indicate numerical problems.
For example, severe artifacts arise when the nonbonded energy interactions
are truncated abruptly. This is because the associated force terms rise violently
at the truncation boundary. Improved techniques for smoothly handling the non-
bonded terms are necessary [1220], as described in Chapter 10. The alternative
approach described also in that chapter is to forgo cutoffs and compute all the
nonbonded terms by fast summation techniques such as multipole or Ewald
[142, 486]. Fortunately, such schemes approach linear, rather than quadratic,
dependence on system size.
13.3.6 High-Speed Implementations
High-speed computers are essential for performing the computationally-intensive
MD simulations [143, 658]. The dynamics of condensed systems was simulated
much earlier this century, but macromolecular applications only gained momen-
tum in the mid-to-late 1980s with the advent of high-speed computing. There are
many reasons why this lag occurred, and the issue at heart is best captured by the
following statement by Frauenfelder and Wolynes [423]:
Whatever complexity means, most people agree that biological
systems have it.
Indeed, the energy landscape is complex for biomolecules [422, 1386, 1387].
The various contacts — be they hydrogen bonds, disulfide bonds, or noncovalent
interactions like stacking and favorable electrostatics — are difficult to predict
apriori. Thus, the multidimensional potential energy surface that governs biomo-
lecular structure has many maxima, minima, and saddle points. The distributions
about each favorable or unfavorable state are highly anisotropic, with the width
depending on the entropy associated with that state.
Biomolecules are also asymmetric in comparison to simple systems, such as
homogeneous liquid clusters, which were successfully simulated much earlier.
Certainly, there are symmetries in many aspects of protein and nucleic acid struc-
ture (e.g., many proteins are dimers, and the “ideal” DNA double helix has an
axis of symmetry), but in realistic environments there are many sequence-specific
motifs and binding interactions with other biomolecules in the environment that
induce local structural variations in macromolecules. These local trends can
produce profound global effects.
The motion of biomolecules is also more complex than that of small or homo-
geneous systems. The collective motion is a superposition of many fundamental
motions that characterize the dynamics of a biomolecule: bond stretches, an-
gle bends, torsions, and combinations of those “normal modes” (see Chapter 9
444 13. Molecular Dynamics: Basics
10
−3
10
−2
10
−1
10
0
10
1
9.12
9.13
9.14
9.15
9.16
9.17
9.18
Convergence of Butane
,
s End−to−End Distance
Δ t (fs)
End−to−end distance (Angstrom)
3−timestep integration
1−timestep integration
10
−3
10
−2
10
−1
10
0
9.127
9.128
9.129
9.130
C1
C4
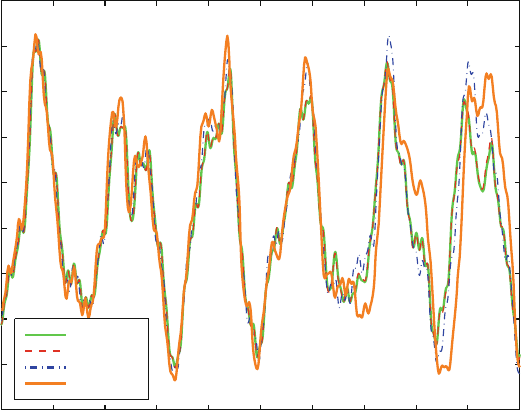
Figure 13.5. Average butane end-to-end distance for single (dashed line) and triple (solid
line) timestep protocols (note logarithmic scale) as computed over 1 ps (see Figure 13.4).
In the latter, the timestep used to define the data point is the outermost timestep Δt (the
interval of updating the nonbonded forces), with the two smaller values used as Δt/2
and Δt/4 (for updating the dihedral-angle terms and the bond-length and angle terms,
respectively).
for definitions and examples). Although these components have associated fre-
quencies of disparate magnitudes, the overall motion of a biomolecule is highly
cooperative. That is, small local fluctuations can trigger a chain of events that will
lead to a large global rearrangement. Energy transfer among vibrational modes
may also be involved.
In addition to the overall asymmetry and complexity (of energy landscape
and motion), the solvent and ions in the environment influence the structure
and dynamics of biomolecules profoundly. Solvent molecules surround and inter-
penetrate proteins and nucleic acids and damp many characteristic motions that
might be present in vacuum. Similarly, ions (sodium, calcium, magnesium, etc.)
influence structure and dynamics significantly, especially of the polyelectrolytic
nucleic acids [850, for example]. Thus, for physical reliability, these long-range
electrostatic interactions are essential to consider in MD simulations of macro-
molecules. This has proven especially important for stabilizing DNA simulations
and accurately describing kinetic processes involving conformational transitions
of DNA [219,220].

13.3. The Basics: An Overview 445
13.3.7 Analysis and Visualization
Careful analysis of the results and visualization techniques are also essential
components of biomolecular simulations today. With the increasing ease and ac-
cessibility of generating computer trajectories, the challenge remains of carefully
analyzing the voluminous data to distill the essential findings. While many scalar
and vector functions can be computed from a long series of configurations, under-
standing the dynamics behavior requires a combination of sophisticated analysis
and chemical/biological intuition.
Molecular graphics techniques were more of a problem in the early days of
molecular mechanics than they are now. Before the surge of graphics innovations,
researchers relied more heavily on mechanical models and physical sense (see
related comment in this book’s Preface).
These days, molecular modeling software and analysis tools have become a
large industry. Many sophisticated and useful tools are available to both ex-
perimentalists and computational/theoretical chemists. The dazzling capability
of computer graphics today to render and animate a large, complex three-
dimensional image — often so “real” in appearance that the source may be
obscured — has certainly made biological interpretation much easier, but one
still has to know exactly where, and for what, to look!
13.3.8 Reliable Numerical Integration
To increase the reliability of macromolecular simulations, special care is needed
to formulate efficient numerical procedures for generating dynamic trajectories.
Mathematically, there are classes of methods for conservative Hamiltonian sys-
tems termed symplectic that possess favorable numerical properties in theory and
practice [1090]. In particular, these schemes preserve volumes in phase space (as
measured rigorously by the Jacobian of the transformation from one set of coordi-
nates and momenta to the next). This preservation in turn implies certain physical
invariants for the system.
Another view of symplectic integration is that the computed trajectory (with
associated Hamiltonian H(X
n
,V
n
)) remains close in time to the solution of a
nearby Hamiltonian
H. This proximity is rigorously defined as a trajectory whose
energy is order O(Δt)
p
away from the initial value of the true Hamiltonian H:
H ≡
1
2
(V
0
)
T
M(V
0
)+E(X
0
) ,
where p is the order of the integrator, and X
n
and V
n
are the collective position
and velocity vectors at time nΔt. This symplectic property translates to good
long-time behavior in practice: small fluctuations about the initial (conserved in
theory) value of H, and no systematic drift in energy, as might be realized by a
nonsymplectic method.
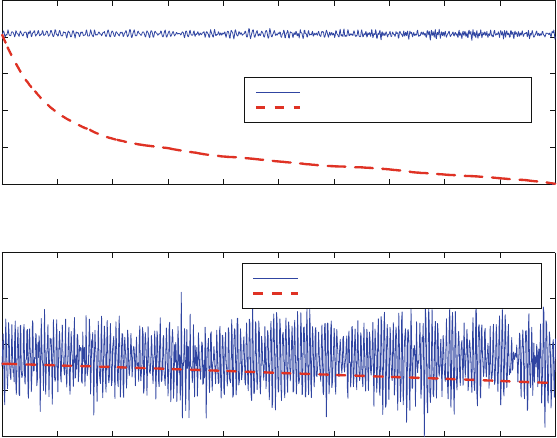
Figure 13.6 illustrates this favorable symplectic behavior for a simple nonlinear
system, a cluster of four water molecules, integrated by Verlet and by the classical
446 13. Molecular Dynamics: Basics
0 2 4 6 8 10 12 14 16 18 20
−27.5
−27
−26.5
−26
−25.5
−25
Velocity Verlet, Δ
t
= 1 fs
Runge−Kutta 4, Δ
t
= 1 fs
0 2 4 6 8 10 12 14 16 18 20
−25.464
−25.4635
−25.463
−25.4625
−25.462
Velocity Verlet, Δ
t
= 0.1 fs
Runge−Kutta 4, Δ
t
= 0.1 fs
Time (ps)
Total Energy (Kcal/mol)
Figure 13.6. Energy evolution of a water tetramer simulated by the symplectic Verlet
scheme (solid line) versus the nonsymplectic Runge-Kutta integrator (dashed line) at two
timesteps Δt (0.1 and 1 fs). Note the different energy scales of the plots.
fourth-order (nonsymplectic) Runge-Kutta method. A clear damping trend (i.e.,
decrease of energy with time) is seen by the latter, non-symplectic integrator,
especially at the larger timestep (top).
While symplectic methods can follow kinetics accurately (assuming the
force field is adequate), other approaches as discussed in the next chapter —
Langevin and Brownian dynamics — can be effective for thermodynamic sam-
pling. Though additional terms are added (e.g., random thermal forces), enhanced
sampling made possible by larger timesteps can yield more information on the
thermally-accessible configuration space.
13.3.9 Computational Complexity
Intensive Requirements
Trimming the computational time of MD simulations remains a challenge [1110].
As seen from Table 1.2 of Chapter 1, the all-atom molecular models used for
biomolecules are quite large (thousands of atoms). The energy and derivative
computations required at each dynamic step are expensive and limit the total
time that can be simulated: typical timesteps (1 fs) imply one million integration
steps per nanosecond. Interestingly, Table 1.2 also shows our tendency to increase
13.3. The Basics: An Overview 447
system size rather than the simulation time. This can be explained, in part, by the
desire to model more complex and realistic systems and by the timespan/sampling
limitation of MD. Dynamic motions over substantially longer times, as well as
larger spatial scales, can only be approximated through macroscopic models of
proteins and DNA (see [1119] for example).
To reduce computational time of MD simulations, two basic routes can be taken
and combined: reducing the work per timestep, and/or increasing the integration
timestep, Δt. In the latter case, the parameter we aim to lengthen is that associated
with long-range force calculations, that is, the ‘outermost timestep’ in multiple-
timestep (MTS) methods.
Less Work Per Step
Reducing the work per step can be achieved with various protocols used in
practice: employing cutoff ranges for the nonbonded interactions, updating the
nonbonded pairlist infrequently (i.e., less often than every outer timestep), and
reducing the number of degrees of freedom by constraining certain molecular
regions (e.g., protein residues revealed by experiment not to be important for a
certain localized transition).
Larger Timesteps: Accuracy vs. Stability
The maximum value for the outer timestep that can be successfully used is limited
by both accuracy and stability considerations, as will be detailed below. Essen-
tially, instability of a trajectory is easily detected when coordinates and energies
grow uncontrollably. This often signals that the timestep is too large. However,
unless the timestep is far too large, this instability may only be apparent from a
trajectory of length of hundreds of picoseconds or longer. The guidelines for sta-
bility for a given integrator are often derived from linear analysis (i.e., theoretical
analysis for a harmonic oscillator) [866,967, 1437, for example]. In practice, the
timestep must typically be smaller than this limit for complex nonlinear problems
to ensure stability as well as reasonable accuracy.
Accuracy limits the timestep Δt in the sense that the resolution produced
must be adequate for the processes being followed and the measurements tar-
geted. Thus, a simulation may be stable but the timestep too large to capture
high-frequency fluctuations accurately.
Closely related to stability and accuracy are resonance artifacts. This topic of
recent interest will be discussed separately.
Techniques for increasing the timestep range from constraining the fastest
degrees of freedom via algebraic constraints to using multiple-timestep (MTS)
schemes to formulating rigid-body dynamics. For recent reviews, see [1110,
1118], for example.
Constraining Fastest Motions
The technique of constrained dynamics has been used for a long time to enforce
rigidity of bond lengths [868,1079,1291,1292]. This approach helps increase the
448 13. Molecular Dynamics: Basics
timestep by a typical factor of two (e.g., from 1 to 2 fs), with similar gains in
the computational time. Freezing bond angles, however, alters the overall motion
substantially [1296]. See the last section of this chapter.
Splitting Forces in MTS Schemes
In MTS schemes, we divide the force components into classes and update the
associated forces at timesteps appropriate for the class. Three-class partitioning
schemes are common in biomolecular simulations, though more classes can be
used for added accuracy/efficiency.
For example, a simple partitioning based on potential-energy components
[1349] can place bond-length and bond-angleterms into the fastest class, dihedral-
angle terms into the medium class, and the remaining forces into the slow-force
class. Incorporating distance classes (e.g., interactions within 6
˚
A into the
medium-force class) involves more complex bookkeeping work for the non-
bonded pairlist generation but often works better. The numerical details of how
these different updates are merged affect the stability of the trajectory significantly
[94,134]; see the separate section on MTS methods in the next chapter for details.
To illustrate a simple MTS scheme, consider using three timesteps for the bu-
tane system analyzed above for computing the end-to-end distance. We set the
three values as Δτ,2Δτ ,and4Δτ ≡ Δt; thus a 3-timestep integrator associated
with the outer timestep of Δt =2fs has an inner timestep of 0.5 fs and a medium
timestep of 1 fs. Bond-length and angle terms are updated every Δτ; dihedral-
angle terms are updated every 2Δτ; and nonbonded interactions are recalculated
every 4Δτ for this butane model. This partitioning should thus save work for this
MTS protocol {Δτ,2Δτ, 4Δτ} relative to the single-timestep integrator at Δτ.
The data shown in Figure 13.5 for the average end-to-end distance of bu-
tane compares values obtained from single-timestep methods to those obtained
by Verlet-based MTS simulations. We see that the accuracy is determined
by the innermost timestep. This is precisely the advantage exploited in MTS
schemes: same accuracy at less computation (under the appropriate comparison)
[579,1448].
For the small butane system, the MTS force splitting is not computation-
ally advantageous because the nonbonded interactions represent a small fraction
of the total force-evaluation work. However, for biomolecules, non-negligible
speedups (e.g., factor of 2 or more) can be reaped with respect to single-timestep
simulations performed at the inner timestep (Δτ) used in the MTS protocol.
13.4 The Verlet Algorithm
One of the simplest and best family of integrators for biomolecular dynamics is
the leapfrog/ Verlet/ St¨ormer group. The basic scheme was derived as a truncation
of a higher-order method used by St¨ormer in 1907 [1227] to integrate trajectories
of electrons. The scheme was adapted by Verlet 60 years later [1302]. A favorable
13.4. The Verlet Algorithm 449
numerical property of this family is exceptional stability over long times (recall
the example in Figure 13.6, where the Verlet trajectory shows favorable energy
conservation compared to the Runge-Kutta trajectory).
Stability far more than accuracy limits the timestep in simulations of biomo-
lecular motion governed by approximate force fields. Programming ease is a
boon for weary simulators who must wade through thousands of lines of molec-
ular mechanics and dynamics programs. Far from mere convenience, simplicity
in the integration also facilitates the implementation of variations in the basic
method (e.g., constrained dynamics, extensions to various statistical ensembles)
and the merging of integration schemes with other program components (e.g., fast
electrostatics by the particle-mesh Ewald method).
For both these practical and theoretical reasons, it has become very difficult
for alternative schemes — such as higher-order and implicit discretizations —
to compete with Verlet in popularity [1110]. For the same reason, the single-
timestep Verlet method is still more popular in general than MTS variants (as
introduced in Subsection 13.3.9 and discussed in detail in the next chapter), but
this is likely to change as comprehensive and reliable MTS routines for various
statistical ensembles enter into the popular biomolecular dynamics packages.
Not surprisingly, generalizations of the Verlet method have been applied to
other formulations, such as constrained and stochastic dynamics. The Verlet
scheme is also the basis of the common force-splitting MTS methods and for
approaches using various thermodynamic ensembles.
Below we describe how the scheme is formulated and derive its leapfrog,
velocity, and position variants.
13.4.1 Position and Velocity Propagation
Iterative Recipe
To derive the Verlet propagation scheme for Newtonian dynamics, we write the
equation of motion, eq. (13.3), as
M
¨
X(t)=F (X(t)) (13.7)
where
F (X(t)) = −∇E(X(t)) . (13.8)
We also use the symbol
F (the acceleration, or the force scaled by the inverse-
mass matrix) to simplify the formulas below, where
F (X(t)) = M
−1
F (X(t)) = −M
−1
∇E(X(t)) . (13.9)
In a numerical discretization, a timestep Δt is chosen and the continuous variables
X(t) and V (t) are approximated by values at discrete time intervals nΔt for
n =0, 1, 2,... ,denoted as X
n
and V
n
. Below we also use the notation F
n
as
short hand for the force at time nΔt, namely F(X
n
). Similarly,
F
n
represents
the numerical estimate to the acceleration at time nΔt:
F (X
n
).

450 13. Molecular Dynamics: Basics
An iterative formula is then chosen to define recursively the positions and
velocities of the molecular system. If the formula defines the solution at timestep
n +1in terms of quantities determined at previous iterates, as in
V
n+1
= V
n
+Δt
F
n
,
the discretization method is explicit. If instead the new solution is defined
implicitly, in terms of known and unknown quantities, as in
V
n+1
= V
n
+Δt
F
n+1
,
the method is implicit. The explicit versions generally involve simple algorithms
that (for propagation only) use modest memory, while implicit methods involve
more complex algorithms but may enjoy enhanced stability properties.
Position Update
The original Verlet algorithm [1302] describes an update for trajectory positions,
as follows:
X
n+1
=2X
n
− X
n−1
+Δt
2
F
n
. (13.10)
This is a two-step propagation scheme since positions from two prior steps (n and
n−1) must be stored. A one-step scheme — preferable in terms of storage — can
be obtained by using V
n
instead of X
n−1
as a variable on the left-hand side (see
below).
The Verlet algorithm is fourth-order accurate in position. To see this, use the
Taylor expansion around X(t) to obtain:
X(t +Δt)+X(t − Δt)
= X(t)+ΔtV(t)+
Δt
2
2
F (X(t)) +
Δt
3
6
¨
V (t)+O(Δt
4
)
+ X(t) − ΔtV(t)+
Δt
2
2
F (X(t)) −
Δt
3
6
¨
V (t)+O(Δt
4
)
=2X(t)+Δt
2
F (X(t)) + O(Δt
4
) . (13.11)
Velocity Update
The flexibility in defining compatible velocity formulas from the Verlet position
propagation formula (eq. (13.10)) has led to Verlet variants. These variants rely
on a velocity formula consistent with eq. (13.10).
We derive such a formula by subtracting the Taylor expansion for X(t − Δt)
from that for X(t +Δt), instead of adding as done above, to obtain:
X(t +Δt) −X(t − Δt)
2Δt
= V (t)+O(Δt
2
) . (13.12)
This definition can be written as the central difference approximation
V
n
=(X
n+1
− X
n−1
)/(2Δt) . (13.13)