Bhushan B. Handbook of Micro/Nano Tribology, Second Edition
Подождите немного. Документ загружается.


© 1999 by CRC Press LLC
Other studies have concentrated not on the formation of adhesive bonds, but on the process by which
these bonds are broken as two surfaces are separated. The failure of adhesive bonds is a complicated
phenomenon that involves both interfacial attraction and kinetic effects. Energy dissipation mechanisms
play a dominant role in the function of adhesives because the mechanical energy required to break a
bond, G, can be 10
4
times greater than the reversible work of adhesion, W. Thus, the excess work, G –
W, is dissipated during rupture of the adhesive.
Baljon and Robbins (1996, 1997) used MD simulations to follow the movement of individual atoms
and energy dissipation during the rupture of a thin adhesive film. The model system contained two rigid
solid walls joined by a thin adhesive film. In separate studies, films composed of linear-chain molecules
of between 2 and 32 monomers (Figure 11.15) were investigated. The monomers interacted via a trun-
cated LJ potential and adjacent monomers along each chain were also coupled through an attractive
potential that prevented chain crossing and breaking (Kremer and Grest, 1990). The rigid solid walls
consisted of two (111) planes of an fcc lattice. The wall atoms were coupled to lattice sites by stiff springs
and their nearest-neighbor spacing was fixed at 80% of the equilibrium monomer spacing. The temper-
ature was kept constant by coupling the wall atoms to a heat bath. For the results discussed here, the
chains were composed of 16 monomers, the adhesive contained 2048 monomers, and each wall consisted
of 800 atoms. Rupture was simulated by separating the walls with a uniform velocity. The particle motions,
forces, potential and kinetic energies, and heat flow toward the bath were all monitored throughout the
course of the simulation.*
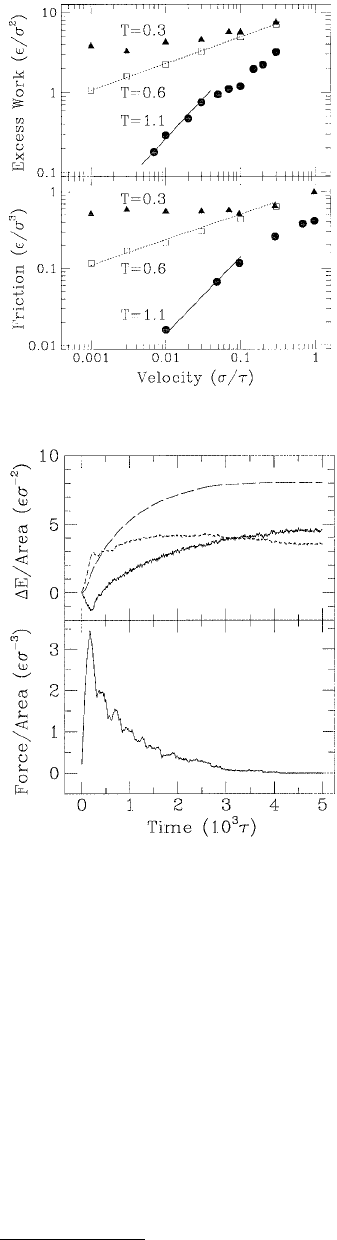
The excess of work G – 2γ (γ is the surface tension) as a function of rupture velocity and the shear
response (friction) vs. shear velocity were examined at three different temperatures (Figure 11.16). The
excess work and the shear response exhibited different behavior in each temperature regime. In the low-
FIGURE 11.15 Snapshots of a glassy film during rupture
at ν = 0.003σ/τ, where σ and τ are characteristic length
and timescales of interaction. The 800 atoms in each wall
are black, and monomers of the 128 chains are colored.
Different colors were used to make the chains forming the
bridges visible. The vertical direction corresponds to the
z-direction and periodic boundary conditions were applied
in the xy plane. Only a thin cross section of the film is
shown in the second panel from the top. (From Baljon
A. R. C. and Robbins, M. O. (1997), Mater. Res. Soc. Bull.
22, 22–26. With permission.)*
* Color reproduction follows page 16.
© 1999 by CRC Press LLC
temperature glassy state, the excess work and friction approached constant values at low velocities, v.
These limits correspond to the amount of adhesion hysteresis and static friction, respectively. Just above
the glass-transition temperature both excess work and friction increased as v
x
, where x = ⅓. At lower
velocities and higher temperatures, there was a newtonian regime in which both quantities increased
linearly with velocity.
Above the glass-transition temperature, rupture produced smooth viscous flows in the film that
resulted in a correspondence between the excess work and the shear stress. In addition, both flow velocities
and dissipation approached zero at low velocities. In contrast, there was no correspondence between the
excess work and the shear stress in glassy films. The shear was confined to the region between the film
and the wall, and rupture occurred in the film through a sequence of rapid structural rearrangements.
As the glassy film was separated, the film deformed elastically, and the force needed to separate the
walls and the distances between layers increased linearly with time (Figure 11.17). Eventually, a threshold
layer separation was reached and the film became unstable against density fluctuations. Small cavities
formed and grew, allowing the remainder of the film to relax to its original density. The interlayer
separation where cavities formed was found to be independent of chain length and, thus, depended only
FIGURE 11.16 Excess work as a function of rupture velocity
(upper panel) and mean frictional force on the tip wall as a
function of shear velocity in the (100) direction at zero pressure
(lower panel). Lines with slope
⅓ (dashed) and 1 (solid) indi-
cate the power law scaling observed at T = 0.6 ε and 1.1 ε,
respectively. (From Baljon, A. R. C. and Robbins, M. O. (1996),
Science 271, 482–484. With permission.)
FIGURE 11.17 Time dependence of energy changes ∆E
(upper panel) and the force on the walls (lower panel) during
rupture of the glassy film shown in Figure 11.15. Lines in the
upper panel indicate the external work (long dashes), the
potential energy increase (short dashes), and the total heat
flow (solid). Because the mean kinetic energy remains con-
stant at fixed temperature, the work equals the sum of the
potential energy change and heat flow. (From Baljon, A. R. C.
and Robbins, M. O. (1996), Science 271, 482–484. With permis-
sion.)

© 1999 by CRC Press LLC
on the force between individual monomers. Each time the internal stress in the film exceeded the local
yield stress, further increases in the wall separation resulted in sudden structural rearrangements of the
film (Figure 11.15, second panel from top). In the latter stages of rupture, the cavities coalesced
(Figure 11.15, second panel from bottom). The lengths of the bridges connecting the walls grew to nearly
that of a fully stretched chain before one end of the chain pulled free and collapsed onto the opposite
surface (Figure 11.15, bottom panel). The final surfaces were very rough. Energy stored in the excess
surface area was part of the unrecoverable work, which was gradually converted to heat as the surface
annealed.
By examining hysteresis loops, the authors determined that the excess work was dissipated evenly
among cavitation, plastic yield, and bridge rupture. All of these processes dissipated more energy with
increased film thickness and chain length. The adhesive energy G for glassy films was approximately twice
the reversible work.
In a similar type of study, Streitz and Mintmire (1996) studied the elastic and yield response of bulk
and thin-film α-alumina. The technological importance of this material at metal–metal oxide interfaces
was the driving force behind its selection. A thin film of α-alumina was constructed from a slab ten layers
thick of α-alumina (about 25 Å thick) with two free (0001) surfaces. Periodic boundary conditions were
applied in the plane of the free surfaces, but not normal to the surfaces. The forces between atoms were
modeled using a variable-charge electrostatic model plus an EAM potential (ES+ method) (Streitz and
Mintmire, 1994). The film was equilibrated; then a sequence of strains was applied by moving the
outermost layers of the film a specified amount normal to the free surfaces. The remainder of the atoms
were allowed to relax, and at each subsequent increment of total strain the internal atoms were allowed
to reach equilibrium.
Streitz and Mintmire (1996) calculated a stress–strain curve for the application of compressive and
tensile strains to both the bulk and thin-film systems. The slab responded elastically during initial loading
and unloading, and the appropriate elastic constant could be calculated from these data. In addition, the
variation of the elastic constant c
33
was also examined as a function of strain. In general, c
33
(the modulus)
increased and the material became stiffer for compressive strains, while for tensile strains the modulus
decreased and the material became softer. For the bulk system, c
33
varied approximately linearly with
strain, while marked deviations from linearity were apparent for the thin film. The value of c
33
at zero
strain was calculated to be 498 GPa for the thin film, in close agreement with the bulk calculation that
yielded 509 GPa. Last, a theoretical yield stress of 44.5 GPa was calculated. This value is not far outside
the theoretically determined range of 35 to 40 GPa. Thus, the authors concluded that the ES+ method
is capable of describing the elastic response of α-alumina in situations that are far from thermodynamic
equilibrium. In addition, these simulations showed that the elastic response of the α-alumina as a thin
film might differ substantially from the elastic behavior of a bulk crystal. This conclusion is quite relevant
in view of the prominent use of α-alumina as a coating.
11.4 Lubrication at the Nanometer Scale:
Behavior of Thin Films
Experiments have shown that the properties of fluids confined between solid surfaces are drastically
altered as the separation between the solid surfaces decreased (Horn and Israelachvili, 1981; Chan and
Horn, 1985; Gee et al., 1990). For instance, at separations of a few molecular diameters, liquid viscosities
increase by several orders of magnitude (Israelachvili et al., 1988; Van Alsten and Granick, 1988). Con-
tinuum hydrodynamic and elastohydrodynamic theories, which have been successful in describing lubri-
cation by micron-thick films, begin to break down when the thickness of the liquid approaches the
thickness of a few molecular diameters. Because an increasing number of applications involve lubricants
in such confined geometries, the need to understand this sort of system through modeling has become
increasingly important.
Molecular dynamics simulations have begun to fill the void created by the breakdown of continuum
theories. These simulations have revealed a number of new phenomena, several of which have explained
© 1999 by CRC Press LLC
experimental observations pertaining to the behavior of confined films. The equilibrium properties of
films of various types, such as spherical molecules, straight-chain alkanes, and branched alkanes confined
between solid parallel walls have been examined. Spherical molecules, for example, have been shown to
order both normal and parallel to the solid walls. Film properties, such as viscosity, have also been
examined. Finally, while macroscopic experiments are consistent with one of the fundamental assump-
tions of newtonian flow, namely, the “no-slip” boundary condition (BC), recent microscopic experiments
are not. (The “no-slip” BC requires that the tangential component of the fluid velocity be equal to that
of the solid surface.) Therefore, flow BCs have been examined using MD simulations (Thompson and
Robbins, 1990a,b; Thompson et al., 1992; Robbins et al., 1993). Some of the more recent work in these
areas is discussed in the following sections.
11.4.1 Equilibrium Properties of Confined Thin Films
A number of groups have studied the equilibrium properties of spherical molecules (interacting through
LJ potentials) confined between solid walls using both Monte Carlo methods (Schoen et al., 1987) and
MD simulations (Bitsanis et al., 1987; Thompson and Robbins, 1990b; Sokol et al., 1992; Diestler et al.,
1993). These studies have demonstrated that, irrespective of the atomic-scale roughness of the pore walls,
when a fluid of spherical particles is placed inside a pore, the fluid layers are layered normal to the pore
walls (Bitsanis et al., 1990).
The typical signature used to identify the ordering of the liquid is the liquid density plotted as a
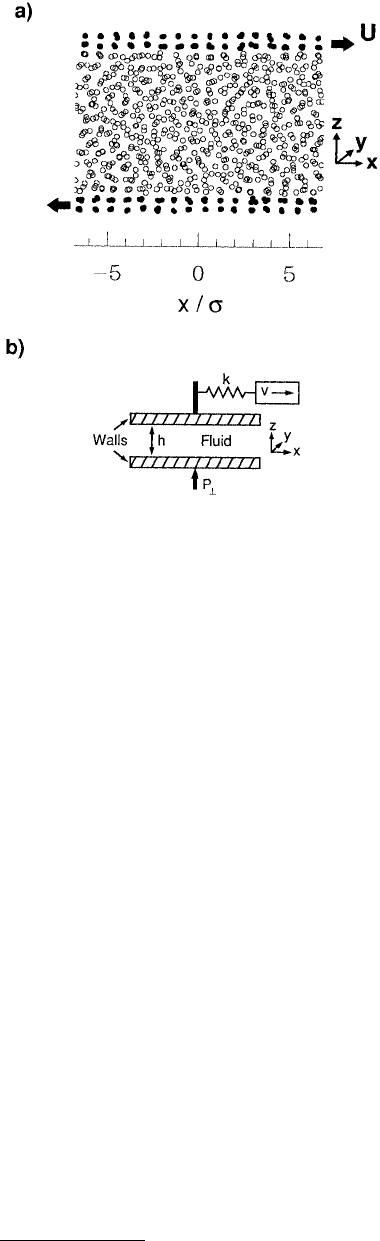
function of distance from the pore walls (termed a density profile). For example, Thompson and Robbins
(1990b) used MD simulations to examine the structure of LJ liquids confined between two solid walls
that consisted of (001) planes of an fcc lattice. The simulation geometry (a slit pore consisting of two
parallel solid surfaces) was chosen to closely resemble an SFA (Figure 11.18a). Oscillations in the calcu-
lated density profiles corresponded to well-defined, liquid layers (Figures 11.19a to c). (In Figure 11.19
the center of the pore corresponds to a z/σ value of zero and the distance z has been normalized by the
characteristic LJ length parameter σ.) In the middle of the pore no oscillations in the density profile were
present; thus, the liquid possessed the unstructured density appropriate for a bulk liquid in this region
of the pore.
FIGURE 11.18 Schematic representation of the sim-
ulation geometry used to model the confinement of
liquids between parallel solid walls. Projections of liquid
particle positions on the xz plane are represented as
open circles and the wall molecules as filled circles in
(a). (From Thompson P. A. and Robbins, M. O. (1990),
Phys. Rev. A 41, 6830–6837. With permission.) There
are 672 fluid atoms and 192 wall atoms. The walls are
moved at a constant velocity U and in opposite direc-
tions along the x-axis. A sketch of a slightly different
simulation geometry is shown in (b). The walls are held
together by a constant load P
⊥
. The upper wall is
attached by a spring to a stage that is moved at constant
velocity v. (From Thompson, P. A. and Robbins, M. O.
(1990), Science 250, 792–794. With permission.) Peri-
odic boundary conditions are imposed in the xy plane.

© 1999 by CRC Press LLC
Normal ordering of the fluid and even a phase transition to a solid structure where layers of the liquid
become “locked” to the solid walls can be induced by increasing the strength of the interaction between
the walls and the fluid (the ratio ε
wf
/ε in Figure 11.19) (Schoen et al., 1989; Thompson and Robbins,
1990b; Sokol et al., 1992). This phenomenon manifests itself as larger oscillations in the density profiles
(Figure 11.19b and c). This effect was also observed when n-alkanes were trapped between structured
walls (Ribarsky and Landman, 1992).
Schoen et al. (1987) were the first to observe that structure in the walls of the pore induces transverse
order (parallel to the walls) in a confined atomic fluid. Using grand canonical MD studies of an atomic
fluid confined between fcc (100) planes of like atoms, they demonstrated that for pore thicknesses of
approximately 1 to 6 atomic diameters, the fluid alternatively freezes and thaws as a function of pore
thickness. The solid formed epitaxially in distorted fcc (100) layers. This epitaxial effect decreased with
increasing pore thickness but persisted indefinitely in the layer nearest to the pore wall. In a related work,
a detailed analysis of the structure of the fluid within a layer, or epitaxial ordering, as a function of wall
densities and wall–fluid interaction strengths was undertaken (Thompson and Robbins, 1990b). For
small ratios of wall-to-liquid well depth (ε
wf
/ε = 0.4), fluid atoms were more likely to sit over gaps in
the adjacent solid layer; however, self-diffusion within this layer was approximately the same as in the
bulk liquid. In other words, although the solid induced order in the adjacent liquid layer, it was not
sufficient to crystallize the liquid layer. Increasing the strength of the wall–fluid interactions by a factor
of 4.5 resulted in epitaxial locking of the first liquid layer to the solid. This epitaxial ordering was
confirmed from an analysis of the two-dimensional structure factors, the spatial probability distribution,
mean-square displacement of the atoms within the layer, and the diffusion within the layer. While
diffusion in the first layer was too small to measure, diffusion in the second layer was approximately half
of its value in the bulk fluid. The second layer of liquid crystallized and became locked to the first “liquid”
layer when the strength of the wall–liquid interaction was increased by approximately an order of
magnitude over its original value. A third layer never crystallized.
The confinement of linear-chain molecules has also been examined by a number of groups (Ribarsky
and Landman, 1992; Thompson et al., 1992; Wang et al., 1993a,b). For example, using a simulation
geometry similar to that shown in Figure 11.18b, Thompson et al. (1992) examined the confinement of
linear-chain molecules between two (111) fcc planes. The linear-chain molecules were modeled via the
bead-spring model, which has been shown to yield realistic dynamics for polymer melts (Kremer and
FIGURE 11.19 Liquid density ρ(z) and the xz component of the micro-
scopic pressure-stress tensor P
xz
as a function of distance between the walls
for a number of wall fluid interaction strengths (ε
wf
/ε). (The wall velocity
U = 0.) The solid line in (d) represents P
xz
averaged within the fluid layers.
All quantities have been normalized using the appropriate variables so that
they are dimensionless. (From Thompson, P. A. and Robbins, M. O. Phys.
Rev. A 41, 6830–6837. With permission.)

© 1999 by CRC Press LLC
Grest, 1990). Adjacent monomers were coupled via an attractive potential and non-nearest-neighbor
monomers interacted via a repulsive, truncated LJ potential.
Confinement of the polymer between solid walls was shown to have a number of effects on the
equilibrium properties of the static polymer films. The film thickness decreased as the normal pressure
on the upper wall increased. At the same time, the degree of layering and in-plane ordering increased,
and the diffusion constant parallel to the walls decreased. In contrast to films of spherical molecules,
where there was a sudden drop in the diffusion constant associated with a phase transition to an fcc
structure, films of chain molecules remained highly disordered and the diffusion constant dropped
steadily as the pressure increased. This indicated the onset of a glassy phase at a pressure below the bulk
transition pressure. This wall-induced glass phase has provided a natural explanation for the dramatic
increases in measured relaxation times and viscosities of thin films (Gee et al., 1990; Van Alsten and
Granick, 1988).
The confinement of n-octane between parallel, crystalline solid walls was examined by Wang et al.
(1993a,b) using MD. A more realistic liquid potential energy function (Jorgensen et al., 1984) was used
and rigid Langmuir–Blodgett (LB) monolayers were used to model the walls of the pore (Hautman and
Klein, 1990). The pore was finite in one direction (typically 2.5 nm long) and made infinite in the other
direction by the application of periodic boundary conditions. In this geometry, liquid exited the pore
and collected as a droplet in the finite direction (Figure 11.20). Liquid vapor from these droplets interacted
with vapors from the other side of the pore via the periodic boundaries (Figure 11.20a to f). The confined
fluid was in equilibrium with the bulk-like droplet at 1 atm and pore widths ranged from 1.0 to 2.4 nm.
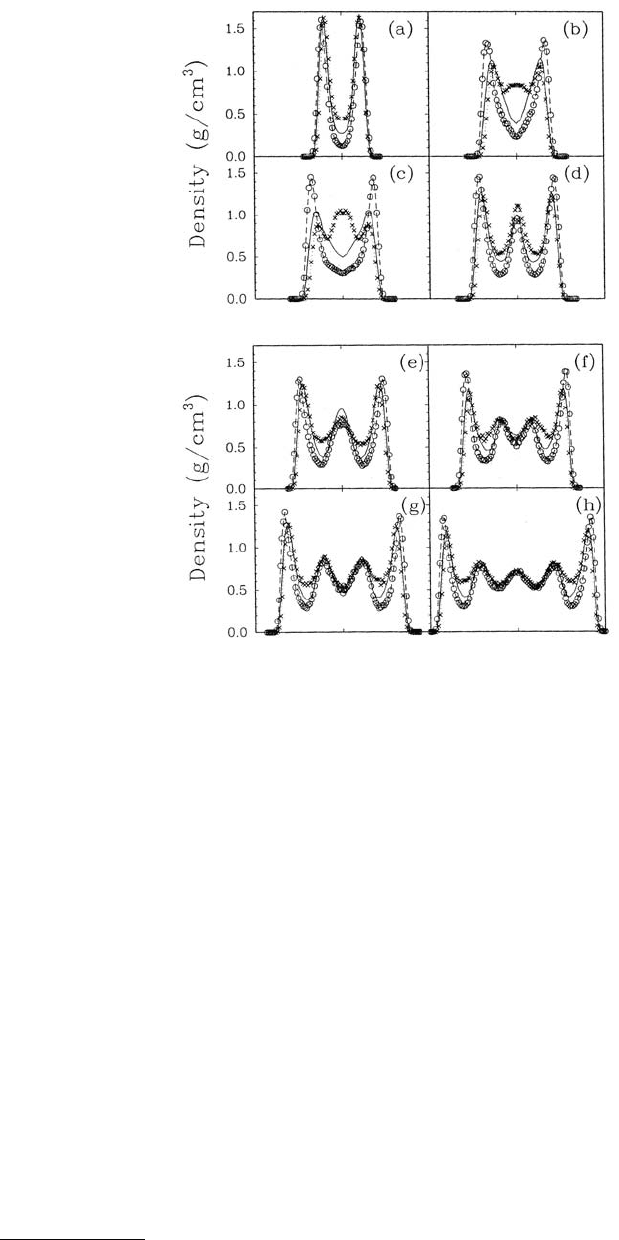
For the smallest pore size examined (1.0 nm) the film formed a layered structure with the molecules
lying parallel to the pore walls (Figure 11.20a). At larger pore widths (Figure 11.20b to f), there was
always a layered structure on each wall surface and more poorly defined layers in the center of the pore,
with the exception of the 1.25-nm pore (Figure 11.20c). In that case, the film ordered so that the alkane
molecules were oriented perpendicular to the walls. The oscillatory nature of the liquid density profiles
(Figures 11.21a to h) confirmed the layered structure of the n-octane films; computed diffusion coeffi-
cients for the films, which were approximately equal to bulk values, confirmed the liquid nature of the
films.
The layering of these films had profound effects on other equilibrium properties. For example, Wang
et al. (1993a) showed that the solvation force of n-octane thin films increased dramatically as the pore
size decreased. Surface force apparatus experiments have also shown that the nature of the film has an
effect on the solvation force. It is well known that linear alkane molecules tend to layer close to a surface.
This layering gives rise to oscillations in the density profile (Christenson et al., 1989). While early
experiments indicated that the surface force oscillations vanish for branched alkanes such as 2-methy-
loctadecane (Israelachvili et al., 1989), more recent experiments (Granick et al., 1995) have shown oscil-
lations in the force profiles of branched hydrocarbon molecules containing a single-pendant methyl group
that are similar to those of linear hydrocarbons. Wang et al. (1993a,b, 1994) carried out MD studies on
confined n-octane and 2-methylheptane and reached a similar conclusion.
In contrast, experimental studies that examined the confinement of highly branched hydrocarbons
such as squalane showed that the surface force oscillations disappear (Granick et al., 1995). In an effort
to shed light on this, Balasubramanian et al. (1996) used both Monte Carlo and MD to examine the
adsorption of linear and branched alkanes on a flat Au(111) surface. In particular, they examined the
adsorption of films of n-hexadecane, three hexadecane isomers (6-pentylundecane, 7,8-dimethyltetrade-
cane, and 2,2,4,4,6,8,8-heptamethylnonane), and squalane. The alkane molecules were modeled using
the united atom approach with an LJ potential used to model the interactions between united atoms.
The alkane–surface interactions were modeled using an external 12-3 potential with the parameters
appropriate for a flat Au(111) substrate (Hautman and Klein, 1989). The heptamethylnonane and
squalane films were investigated using constant-NVT MD simulations. Other films were examined using
configuration biased Monte Carlo (Siepmann and McDonald, 1993a,b; Siepmann and Frenkel, 1992).
The Monte Carlo calculations yielded density profiles for n-hexadecane and 6-pentylundecane that
were nearly identical with experiment and previous simulations. In contrast, the density profiles of the

© 1999 by CRC Press LLC
more highly branched alkanes such as heptamethylnonane and 7,8-dimethyltetradecane exhibited an
additional peak. These peaks arose from methyl branches that could not be accommodated in the first
liquid layer next to the Au surface. That is, the branched hydrocarbons adsorbed in layered structures
with interdigitation of the molecules. For thicker films, the oscillations in the density profiles for hep-
tamethylnonane were out of phase with those for n-hexadecane, in agreement with the experimental
observations (Granick et al., 1993).
Granick et al. (1993) did not observe force oscillations for squalane films confined between mica or
organic monolayers of octadecyltriethoxysilane when the surfaces of the SFA were separated by more
than 18 Å. In contrast, the MD simulations (Balasubramanian et al., 1996) yielded a density profile for
the squalane film that was very similar to the density profile for 7,8-dimethyltetradecane. The most likely
reason for this discrepancy between theory and experiment was the fact that the film was adsorbed, rather
than confined, in the MD simulations.
FIGURE 11.20 Atomic configurations adopted by n-octane when confined between parallel, rigid LB substrates of
various pore widths. The pore widths and liquid temperatures are 1.0 nm and 297 K in (a), 1.6 nm and 297 K in
(b), 1.25 nm and 297 K in (c), 1.25 nm and 250 K in (d), 1.8 nm and 297 K in (e), and 1.8 nm and 250 K in (f).
The light spheres represent fluid (united) atoms and the dark spheres represent substrate atoms. (From Wang, Y.
et al. (1993), J. Phys. Chem. 97, 9013–9021. With permission.)
© 1999 by CRC Press LLC
11.4.2 Behavior of Thin Films under Shear
The behavior of confined spherical and chain molecular films under shear has been examined using both
Monte Carlo methods (Schoen et al., 1987; Curry et al., 1994; Curry and Cushman, 1995a,b) and MD
simulations (see, for example, Schoen et al., 1989; Bitsanis et al., 1990; Bitsanis and Pan, 1993; Sokol
et al., 1992; Diestler et al., 1993; Thompson and Robbins, 1990b; Thompson et al., 1992; Curry et al.,
1994; Curry and Cushman, 1995a,b).
For instance, in an early work Bitsanis et al. (1990) examined the effect of shear on spherical, symmetric
molecules, confined between planar, parallel walls that lacked atomic-scale roughness. Both Couette
(simple shear) and Poiseuille (pressure-driven) flows were examined. The presence of flow had no effect
on the density profile of the liquids as they were identical with the equilibrium density profiles for both
types of flow and all pore widths. Velocity profiles, defined as the velocity of the liquid parallel to the
wall as a function of distance from the center of the pore, should be linear and parabolic for Couette
and Poiseulle flow, respectively, for a homogeneous liquid. In the simulations of Bitsanis et al. (1990) the
velocity profiles for both types of flow deviate from the shape expected for homogeneous flow in the
FIGURE 11.21 Liquid density profiles in the direction normal to the surface of the pore walls as a function of pore
separation. The center of the pore is indicated by the single tick mark on the x-axis. Density profiles for all hydrocarbon
groups within the liquid molecules are represented with a solid line, for methyl groups 1 and 8 with open circles,
and for methylene groups 4 and 5 by crosses. The pore widths are 1.0 nm in (a), 1.2 nm in (b), 1.25 nm in (c),
1.4 nm in (d), 1.6 nm in (e), 1.8 nm in (f), 2.0 nm in (g), and 2.4 nm in (h). All liquid films are at 297 K. (From
Wang, Y. et al. (1993), J. Phys. Chem. 97, 9013–9021. With permission.)
© 1999 by CRC Press LLC
regions occupied by the two outermost fluid monolayers. These outermost layers behaved like a fluid of
very high viscosity.
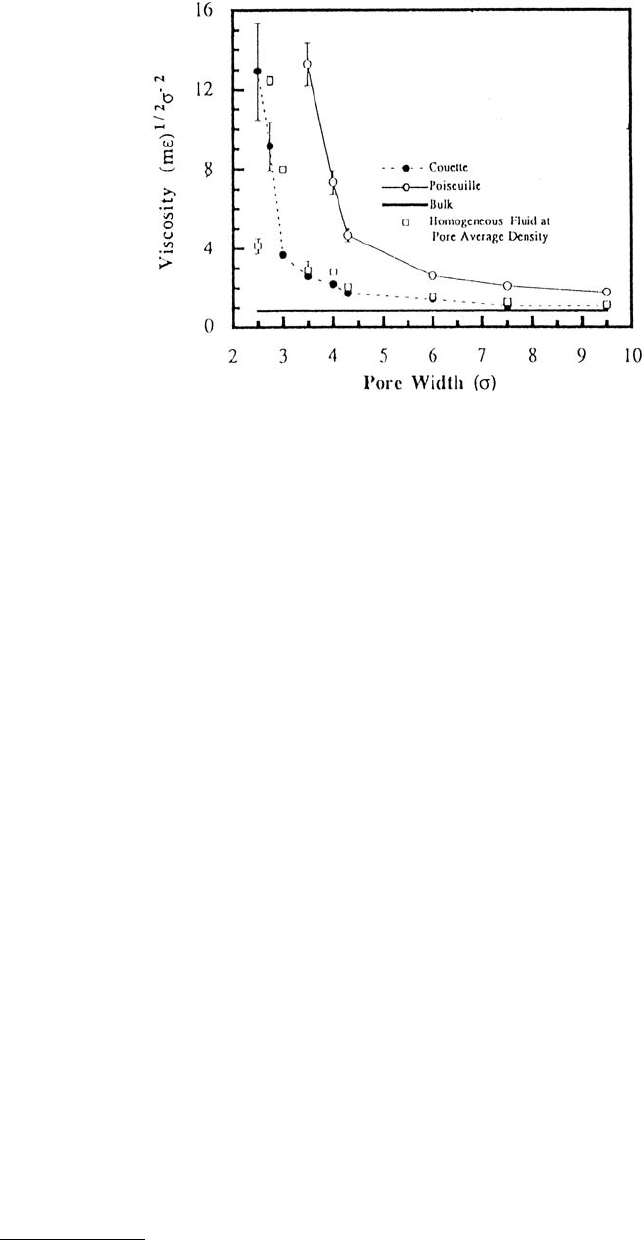
The profoundly different nature of flow in molecularly thin films was further demonstrated by plotting
the effective viscosity vs. pore width (Figure 11.22). For a bulk material the viscosity was independent
of pore size. However, under both types of flow, the viscosity increased slightly as the pore size decreased.
For ultrathin films, the viscosity increased dramatically.
The effect of transverse order on confined thin films under shear, induced by atomic structure in the
walls, was also examined. Schoen et al. (1989) examined the behavior of argon LJ films under shear,
confined between fcc (100) walls, using both grand canonical Monte Carlo and microcanonical MD
simulations. At small wall separations of 2.0 σ, the pore accommodated two commensurate solid-like
layers when the walls were fully out of registry with one another. Shearing caused the stress to increase
linearly as the walls came closer into registry with one another. The number of atoms in the pore remained
essentially constant until a critical stress was reached, at which point continued shearing caused almost
an entire layer of fluid to exit from the pore, thereby decreasing the stress. These results are in agreement
with experimental data, which indicates that solid surfaces slide past one another while separated by a
discrete number of layers. Further, a critical shear stress was required to initiate sliding under appropriate
conditions.
In contrast, with wall separations appropriate for an odd number of fluid layers (3.10 σ and 4.90 σ)
the pore supported either three or five layers when the opposing surfaces were in registry. For the system
with three solid-like layers, the shear stress increased linearly when the system was sheared. As in the
previous case, this trend continued until a critical stress was achieved. Continued strain on the walls
resulted in the drainage of one of the solid layers.
The flow of LJ liquids confined between two solid walls was also examined by Thompson and Robbins
(1990b). In this case, the walls were composed of (001) planes of an fcc lattice. A number of wall and
fluid properties, such as wall–fluid interaction strength, fluid density, and temperature, were examined.
The geometry of the simulations closely resembled the configuration of an SFA (Figure 11.18b). Each
wall atom was attached to a lattice site with a spring to maintain a well-defined solid structure with a
minimum number of solid atoms. The spring constant controls the thermal roughness of the wall and
its responsiveness to the fluid, and it was adjusted so that the mean-square displacement about the lattice
FIGURE 11.22 Effective viscosity for Couette and Poiseuille flows of an LJ liquid as a function of pore width. Pore
width is given in units of the LJ parameter σ (parameters used in this simulation were appropriate for argon). The
pore walls lack atomic-scale roughness. The viscosity of a homogeneous fluid at the pore average density and that
of a bulk liquid are also shown. (From Bitsanis I. et al. (1990), J. Chem. Phys. 93, 3427–3431. With permission.)
© 1999 by CRC Press LLC
sites was less than the Lindemann criterion for melting. The interactions between fluid atoms and between
wall and fluid atoms were modeled by different LJ potentials. Couette flow was simulated by moving the
walls at a constant velocity in opposite directions along the x-axis (Figure 11.18). The heat generated by
the shearing of the liquid was dissipated using a Langevin thermostat. In most of the simulations
performed by Thompson and Robbins (1990b), the fluid density and temperature were indicative of a
compressed liquid about 30% above its melting temperature, with 672 fluid atoms, and either 192 or
352 wall atoms.
A number of interesting phenomena were observed in these simulations. First, the well-defined lattice
structure of the solid walls induced both normal and parallel ordering in the adjacent liquid. The normal
ordering of the films was apparent from the oscillations in the density profiles (Figure 11.19a to c).
Density oscillations in the liquid also induced oscillations in other microscopic quantities normal to the
walls such as the fluid velocity in the flow direction V
x
and the microscopic pressure–stress tensor P
xz
(Figure 11.19d). The position of the peaks in P
xz
corresponded to the peak positions in the liquid density,
while peak positions in dV
x
/dz are between the layers. These observations were contrary to the predictions
of the Navier–Stokes equations. However, it was determined that averaging dV
x
/dz and P
xz
over length
scales that are larger than the molecular length σ produced smoothed quantities that satisfied the
Navier–Stokes equations (smooth line in Figure 11.19d).
While the ordering of the fluid normal to the walls affected fluid flow, two-dimensional ordering of
the liquid parallel to the walls affected the flow more significantly. The velocity of the fluid parallel to
the wall was examined as a function of distance from the wall for a number of wall–fluid interaction
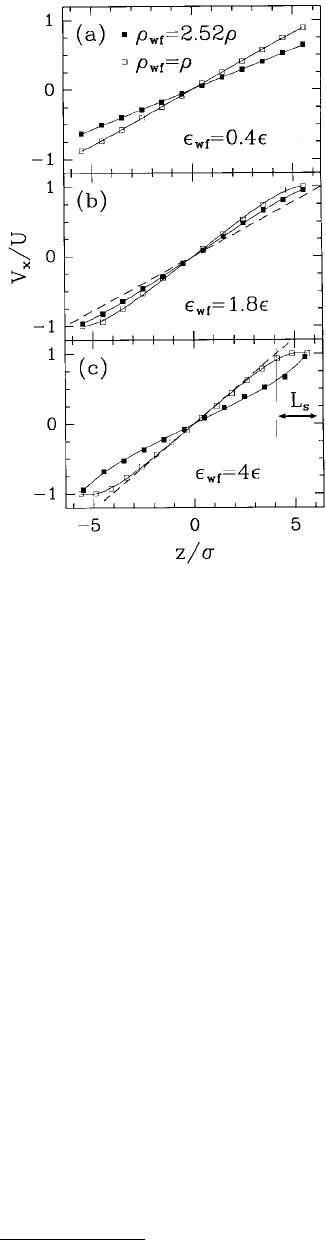
strengths and wall densities (Figure 11.23). Analysis of velocity profiles showed that flow near solid
boundaries is strongly dependent on the strength of the wall–fluid interaction and on wall density. For
instance, when the wall and fluid densities were equal and wall–fluid interactions strengths were small
(ε
wf
/ε = 0.4), the velocity profile was linear with a no-slip BC (Figure 11.23a). As the wall–fluid interaction
strength increased, the magnitude of the liquid velocity in the layers nearest the wall increased; thus, the
FIGURE 11.23 The component of the liquid velocity in the
shearing direction V
x
vs. the position between the pore walls z.
The locations of the first layers of solid atoms on each side of
the fluid correspond to the vertical borders. Data points indicate
averages of V
x
within the layers and the solid lines represent
polynomial fits to these data. The dashed line in (b) represents
the flow expected from hydrodynamics with a no-slip BC. The
liquid velocity has been normalized by the velocity of the walls
and the pore width by the LJ length parameter σ. (From
Thompson, P. A. and Robbins, M. O. (1990), Phys. Rev. A 41,
6830–6837. With permission.)