Kosevich A.M. The crystal lattice
Подождите немного. Документ загружается.

1.2 Motion of a Localized Excitation in a Monatomic Chain
25
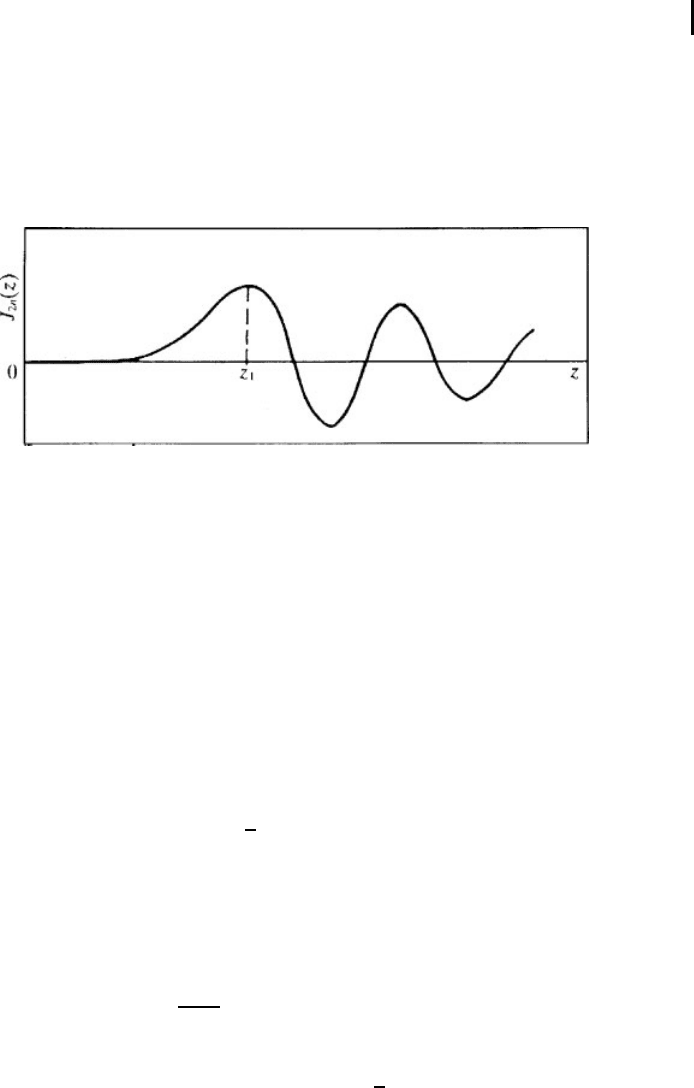
(major) perturbation maximum comes at the point n(n 1) at a time t
1
determined
by the condition ω
m
t
1
≈ 2n (Fig. 1.3). The velocity of motion of a perturb a tion
maximum is na/t
1
≈ (1/2)ω
m
a = s, i. e., it is practically the same as the sound
velocity in a 1D crystal. Thus, the effective velocity of the transfer of the perturbation
is not different from that of the sound velocity, i. e., it is the same as the limiting group
velocity that follows from the d isp ersion relation (1.1.6) or (1.1.9).
Fig. 1.3 The time dependence of the perturbation coming
to the point located at a distance an from the displaced atom
(z = ω
m
t, z
1
= ω
m
t
1
≈ 2n).
Having clarified the role of the dispersion in an excitation signal moving along the
1D discrete chain, we now describe the velocity dispersion in the long-wave con-
tinuous approximation. It is known that competition of the higher dispersion with
nonlinearity is very important in the dynamics of complex media. Hence, deriving a
dynamic equation in partial derivatives for the 1D crystal anew, we take into account
unharmonic terms in the interaction energy of the nearest n eighbors. We assume the
potential energy of a crystal to have the form of a sum
U =
∑
n
ϕ (ξ
n
)=
∑
n
ϕ (u
n
− u
n−1
); ξ
n
= u
n
−u
n−1
, (1.2.4)
so that in the harmonic approximation
ϕ (ξ)=
1
2
ϕ
(0)ξ
2
, ϕ
(0)=α > 0. (1.2.5)
Generalizing the equations of motion of a 1D crystal, we assume the function ϕ(ξ)
to be different from (1.2.5) and reduced to the p arabolic dependence (1.2.5) only at
ξ → 0.
The equation of crystal motion with interactions between nearest neighbors only
has the form
m
d
2
u
n
dt
2
= ϕ
(u
n+1
− u
n
) − ϕ
(u
n
− u
n−1
). (1.2.6)
For small relative displacements, one can take
ϕ
(ξ)=ϕ
(0)ξ +
1
2
ϕ
(0)ξ
2
, (1.2.7)
including only the so-called cubic anharmonicity.
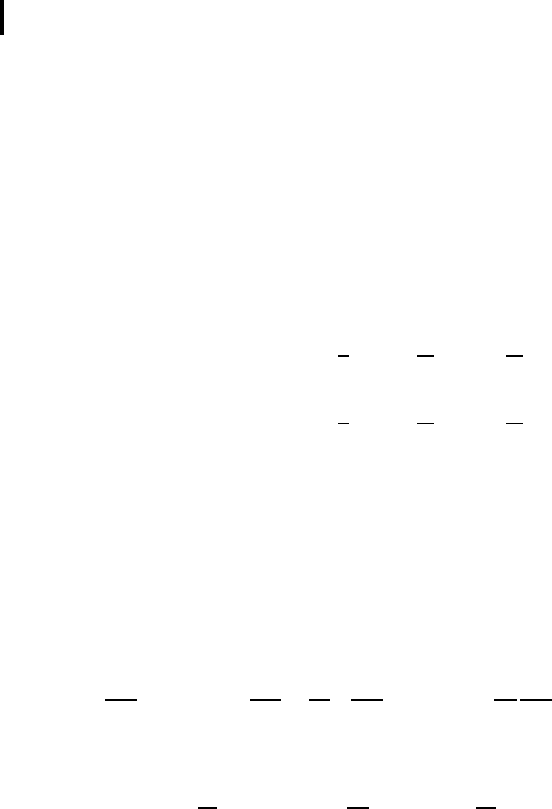
26
1 Mechanics of a One-Dimensional Crystal
We substitute (1.2.7) into (1.2.6) and compare the result with (1.1.1); it is seen that
α = mϕ
(0). Just this relation connects the elements of the force matrix introduced
phenomenologically with the parameter of the interatomic interaction potential.
However, (1.2.6) allows one to avoid the restrictions that arise in considering the
harmonic vibrations by means of (1.1.4). To enable a description of inhomogeneous
crystal states varying weakly in space we consider (1.2.6) in the long-wave approxi-
mation. Assume the characteristic distance ∆x of the change in the field of displace-
ments to be large ( ∆x a). This makes it possible to pass to a continuum treatment,
i. e., to replace the functions of a discrete argument n by the function of a continuous
coordinate x and use the expansions
ξ
n+1
= u
n+1
− u
n
= au
+
1
2
a
2
u
+
1
3!
a
3
u
+
1
4!
a
4
u
;
ξ
n
= u
n
− u
n−1
= au
−
1
2
a
2
u
+
1
3!
a
3
u
−
1
4!
a
4
u
.
(1.2.8)
In (1.2.8), the terms with the fourth-order derivatives remain, since the corresponding
terms in the equations of motion may compete with terms generated by nonlinearity.
The nonlinear terms in (1.2.6) will be calculated to the first nonvanishing approxi-
mation with the lowest orders of the derivatives. Therefore, using (1.2.8), we take
ξ
2
n+1
= a
2
u
2
+ a
3
u
u
, ξ
2
n
= a
2
u
2
− a
3
u
u
. (1.2.9)
Substitute (1.2.8), (1.2.9) into (1.2.7), (1.2.6) to obtain
m
d
2
u
dt
2
= a
2
ϕ
(0)
∂
2
u
∂x
2
+
1
12
a
2
∂
4
u
∂x
4
+ a
3
ϕ
(0)
∂u
∂x
∂
2
u
∂x
2
. (1.2.10)
Introduce the notations
s
2
=
a
2
m
ϕ
(0), B
2
=
a
2
12
s
2
, Λ = −
a
3
m
ϕ
(0), (1.2.11)
and write (1.2.10) as a nonlinear wave equation (the Boussinesq equation):
u
tt
= s
2
u
xx
+ B
2
u
xxxx
− Λ
2
u
x
u
xx
, (1.2.12)
where u
tt
is the second derivative in time; u
x
, u
xx
, u
xxxx
are the derivatives of the
corresponding order with respect to the coordinate x.
Since, when the atoms approach each other their mutual repulsion increases and
when they separate from each other their attraction decreases, it may be assumed
that ϕ
(0) < 0. This was used in introducing the notation of (1.2.11). Besides that,
although a simple relation between the parameters B and s results from the assumption
of interaction of nearest n eighbors only, it is quite natural to regard the coefficient of
∂
4
u/∂x
4
to be positive. Indeed, the dispersion law of harmonic vibration of a 1D

1.2 Motion of a Localized Excitation in a Monatomic Chain
27
crystal is gen erally such that the group velocity dω/dk decreases with increasing k
for small k.
For the equation of motion (1.2.12) the long-wave ( ak 1) dispersion relation for
small (harmonic) vibrations has the form
ω
2
= s
2
k
2
− B
2
k
4
, (1.2.13)
and the group velocity for ak 1 is v = s − (3/2)(B
2
k
2
/s). Thus, the coefficient
discussed should really be positive to provide a decrease in v with k increasing.
An harmonic approximation describes well small crystal perturbations. But in some
cases there arises the necessity to describe the motion of crystal atoms, which is ac-
companied with their large displacements. It is natural to pose the question whether
the motion of crystal atoms is possible in which a strong perturbation will move along
the crystal without changing the form of this perturbation?
If the displacement gradients connected with this perturbation are small there exists
a positive answer to this question w ithin the harmonic approximation. As we have
noted, in this case the atom displacement is described by the linear wave equation
(1.1.14) whose solution is any double-differentiated function depending on the argu-
ment x − st (if the wave runs in the positive direction of the axis x)orx + st (if the
wave runs in the opposite direction). We remind readers that s is the sound velocity.
But if the harmonic approximation is insufficient, the answer to the question posed
is no longer obvious. We study this question using the Boussinesq equation (1.2.12).
Since a transfer of any deformation impulse in a 1D crystal is connected with motion
of a local compression, the analysis of dynamics of the derivative p = u
x
makes sense
supposing that the plot of p(x) has a form similar to Fig. 1.4.
Thus, we find a stationary solution to (1.2.12), moving along the axis x, i. e., a solu-
tion of the form u = u(x − Vt),whereV is an arbitrary parameter (the perturbation
velocity). As the desired function is, actually, a function of one argument ξ = x −Vt
(1.2.12) is transformed into the ordinary differential equation
B
2
u
xxxx
− Λ
2
u
x
u
xx
− γu
xx
= 0, (1.2.14)
where γ = V
2
− s
2
. We introduce new notations α = Λ/B, β = γ/B
2
and r ewrite
(1.2.14) with respect to p:
p
xxx
− α
2
pp
x
− βp
x
= 0. (1.2.15)
We detract from a specific value of the coefficient B given by the definition (1.2.11)
and seek for a formal solution to (1.2.15) for all possible values of β. Integrating
(1.2.15) over x twice, taking into account the boundary conditions at infinity, we ob-
tain
dp
dx
2
−
1
3
α
2
p
3
− βp
2
= 0. (1.2.16)
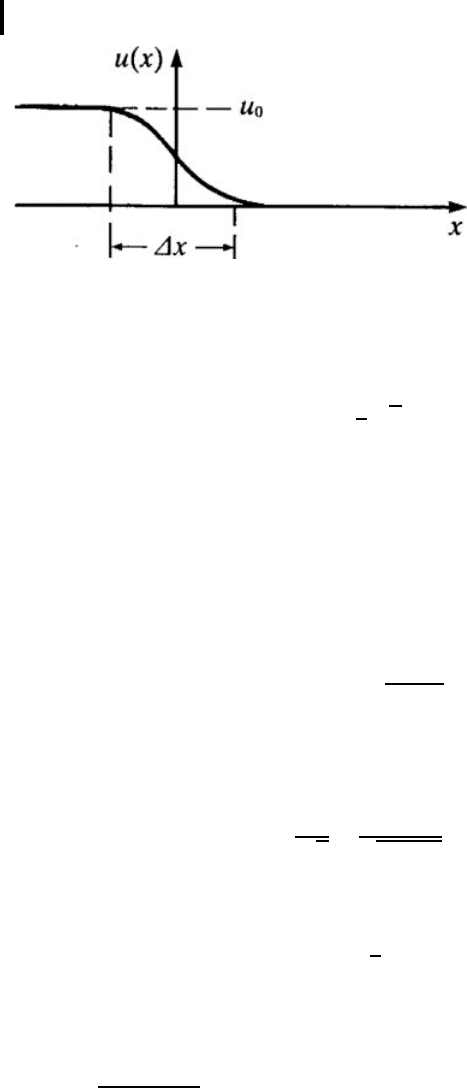
28
1 Mechanics of a One-Dimensional Crystal
Fig. 1.4 Solitary wave of 1D crystal deformation.
Equation (1.2.16) is easily integrated, and we obtain the solution that is interesting
for us
p = −p
0
sech
2
1
2
β(x −Vt)
, (1.2.17)
where p
0
= 3β/α
2
. It follows from the solution (1.2.17) that it exists only for p
0
> 0,
i. e., for V > s .
As the q uantity p has the meaning of 1D crystal strain, the solution (1.2.17) obtained
by us really describes a local compression moving with velocity V along a 1D crystal.
A similar solution for the deformation is called a solitary solution, or a soliton.This
is a singular (isolated) solution to the equation concerned, which may move along the
crystal without changing its form.
Rewrite (1 .2.17)
p = −p
0
sech
2
x −Vt
l
, (1.2.18)
where l is the soliton width determining the transition region ∆x in Fig. 1.4. The value
of l can be found from asymptotes of the solution u ∼ exp(−x/l) of a corresponding
linear equation (obtained from (1.2.14) or (1.2.15) with Λ = 0 ), and it is
l =
1
β
=
B
√
V
2
−s
2
. (1.2.19)
The velocity of the motion of the perturbation concerned is connected with the
amplitude p
0
:
V
2
= s
2
+
1
3
Λ
2
p
0
. (1.2.20)
Thus, the nonlinear differential equation (1.2.14) has the desired solution (1.2.17) only
for the velocity determined by ( 1 .2.20).
In conclusion, it should be noted that substituting a specific value B
2
= a
2
s
2
/12
that follows from (1.2.11) into the for mula for the overall wid th l yields:
l = as/
12(V
2
−s
2
). Thus, the long-wave approximation (l a) is actually
valid only in a narrow interval of velocities V near s.

1.3 Transverse Vibrations of a Linear Chain
29
1.3
Transverse Vibrations of a Linear Chain
We consider a special linear an alog of a simp le crystal lattice assuming the atoms to be
positioned periodically along a certain line in 3D space. Let a be a lattice constant and
n the atom number counted from any point of the chain. We direct the x-axis along
the undeformed straight line chain and denote by v the vector of transverse atom dis-
placements (perpendicular to the x-axis), retaining the notation u for the longitudinal
component of the displacement vector. The great interest in studying the vibrations
of the 1D system proposed is explained by the fact that this problem is an excellent
model of the dynamics of homopolymer molecules.
Keeping in mind possible applications to vibrations of the homopolymer molecules
we restrict ourselves to the long-wave approximations. In the harmonic approximation
the longitudinal and transverse vibrations are independent, and we analyze each form
of motion separately. If the atoms are displaced along the x-axis, the elastic energy is
determined by their relative displacements. The relative displacement of neighboring
atoms is ξ
n
= u
n
− u
n−1
, and, in the nearest-neighbor approximation, the crystal
potential energy equals a sum such as (1.2.4), so that in the harmonic approximation,
it is possible to employ only the expansion (1.2.5). The forces generating the potential
energy (1.2.4), (1.2.5) provide, between the neighboring atoms, a certain analog of
spring coupling with the elasticity coefficients α. Such forces are called central forces.
In going over to the long-wave vibrations with the replacement u
n
→ u(x) can be
effected, the leading term when expanding the difference ξ
n
in powers of a/λ (1.2.8),
where λ is the characteristic wavelength, is proportional to the first derivative of u(x)
with respect to x. The crystal potential energy (1.2.4) then becomes
U =
ϕ
au
(x)
dx
a
, (1.3.1)
and according to (1.2.5) the energy density is
ϕ =
1
2
αa
2
u
2
. (1.3.2)
Equation (1.1.14) with s
2
= αa
2
/m is obtained in a standard way from (1.3.1), (1.3.2).
If the atoms in a linear chain are displaced perpendicular to the x-axis, in the har-
monic approximation central interaction forces do not arise and the crystal energy
depends on the relative rotations of the segments connecting atoms in neighboring
pairs rather than on the relative displacement of neighboring atoms. We assume that
the transverse displacements of all atoms lie in one plane and denote the transverse
displacement in this plane as v
n
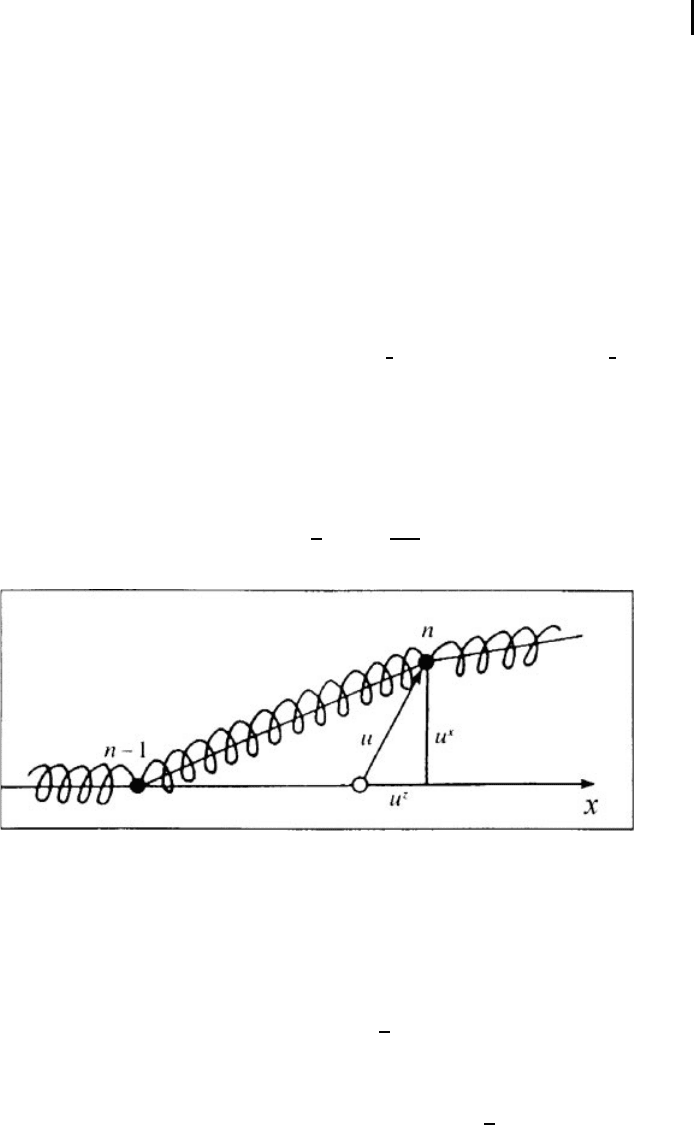
, the angle of similar rotatio n by θ (Fig. 1.5). Then, as
seen from the figure, for small θ one can write for the nearest neighbors
θ
n
=
1
a
(v
n+1
− v
n
) −
1
a
(v
n
−v
n−1
)=
1
a
(v
n+1
+ v
n−1
−2v
n
) .
30
1 Mechanics of a One-Dimensional Crystal
Thus, the crystal energy with noncentral interaction forces of the nearest atom pairs
taken into account will have an additional term
V =
∑
n
ψ(aθ
n
)=
∑
ψ (v
n+1
+ v
n−1
−2v
n
) ,
and, in the harmonic approximation,
ψ(η)=
1
2β
η
2
.
In the long-wave limit, when v
n
is replaced by a continuous function of the coordi-
nate v(x), the leading term of the θ
n
angle expansion in powers of a/λ is proportional
to the second derivative of v(x) with respect to x:
a θ
n
= v
n+1
+ v
n−1
−2v
n
= a
2
∂
2
v
∂x
2
. (1.3.3)
The crystal energy V takes the form of an integral over the whole length of a linear
chain
V =
ψ
a
2
v
(x)
dx
a
,
where the energy density is
ψ =
1
2
βa
4
v
2
. (1.3.4)
Fig. 1.5 Atom configuration in a 1D chain with transverse (bending) vi-
brations.
The potential energy density (1.3.4) leads to the following equation
m
∂
2
v
∂t
2
+ aA
2
∂
4
v
∂x
4
= 0, (1.3.5)
where A
2
= βa
2
/m. Equation (1.3.5) describes the so-called bending vibrations.
Comparing (1.1.14) and (1.3.5) shows that the term with the fourth-order derivative
in (1.3.5) includes a small parameter of the order (a/λ)
2
unavailable in the term with
the second-order derivative in (1.1.14). With such small terms preserved, the linear
approximation may be insufficient. The nonlinearity should be taken into account, in
1.3 Transverse Vibrations of a Linear Chain
31
particular, in the presence of static stretching forces applied to chain ends. Under the
action of such forces there arises a homogeneous longitudinal deformation ∂u/∂x =
0
= const dependent on stretching load, so that it can be large.
In constructing an elementary nonlinear theory of vibrations of the chain concerned,
anharmonicity should be taken into account only in the terms associated with central
forces and the potential energy of small noncentral forces V can be calculated in the
ordinary harmonic approximation.
The main nonlinearity is generated by the anharmo nicities of central forces. If the
nonlinearity is small, the crystal energy can be described by (1.2.4), (1.2.5) having
determined more exactly the relative d istance between neighboring atoms δl
n
, with
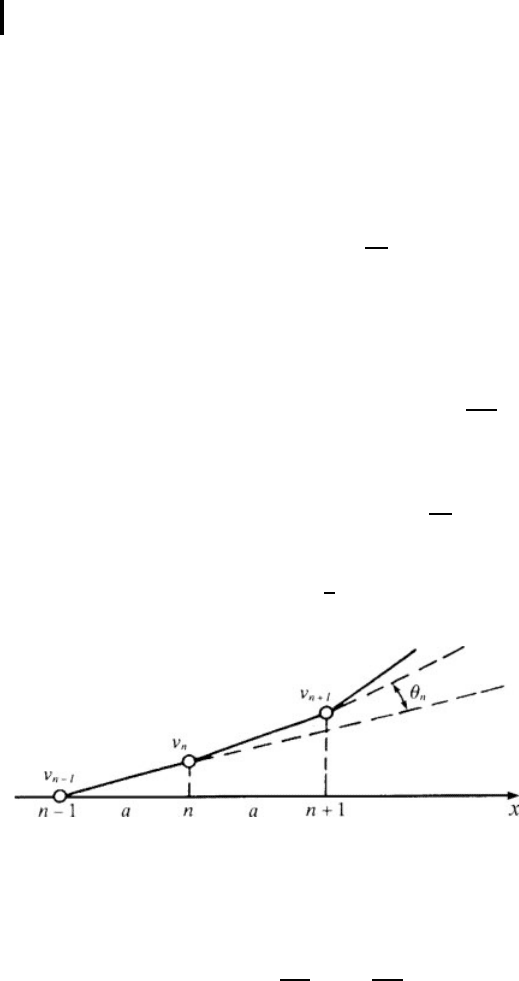
account taken of displacements in a direction perpendicular to the x-axis (Fig. 1.6)
δl
n
=
a + u
z
n
−u
z
n−1
2
+(v
n
−v
n−1
)
2
1
2
− a =
(a + ξ
n
)
2
+ η
2
n
1
2
− a,
where v(u
y
n
, u
z
n
) is the 2D transverse displacement vector, ξ
n
= u
x
n
− u
x
n−1
= u
n
− u
n−1
; η
n
= v
n
− v
n−1
.
To describe the nonlinear bending vibrations of a chain, we use the expression for
δl
n
, written with sufficient accuracy
δl
2
n
= ξ
2
n
+
1
a
ξ
n
η
2
n
+
1
4a
2
η
2
n
η
2
n
. (1.3.6)
Fig. 1.6 A scheme of displacements at transverse 1D chain vibrations
(the displacement of an atom with the number n −1 equals zero).
Comparison of the first two terms on the r.h.s. of (1.3.6) reveals that under bending
vibrations, the value of ξ
n
is commensurate with (l/a)η
2
n
; therefore, we retain the last
term proportional to η
4
n
.
Taking into account the above arguments one should write instead of (1.2.4)
U
∗
=
∑
n
ϕ (δl
n
)=
1
2
α
∑
δl
2
n
,
conserving, for the noncentral interaction energy, the expression
V =
∑
n
ψ (v
n+1
+ v
n−1
−2v
n
)=
∑
n
ψ (η
n+1
+ η
n
)=
1
2
β
∑
n
(η
n+1
+ η
n
)
2
.
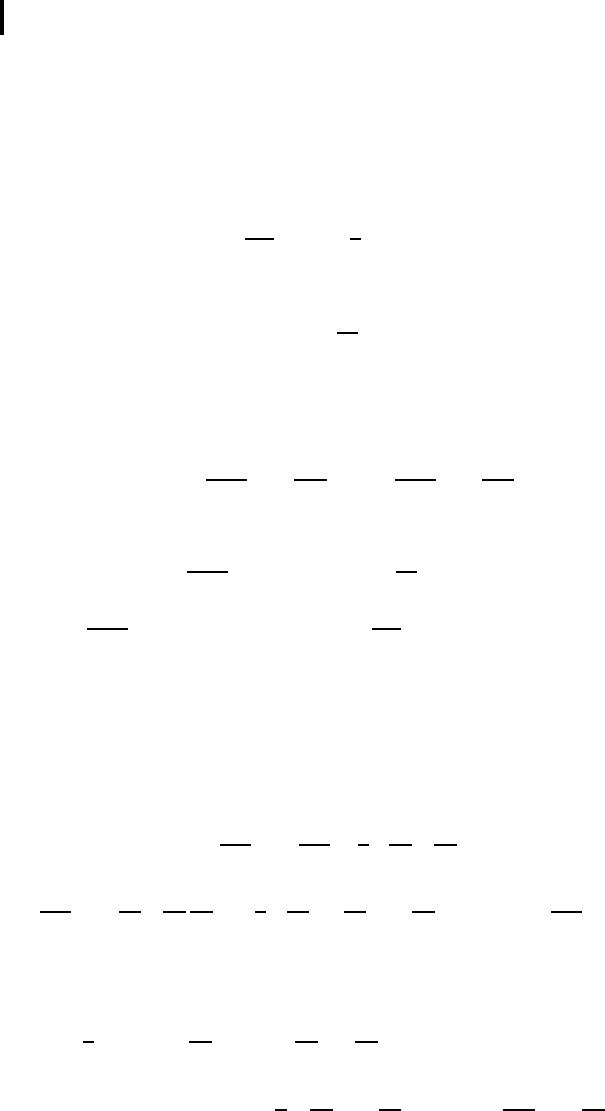
32
1 Mechanics of a One-Dimensional Crystal
It is seen that the total potential energy of the chain W in the nonlinear approxima-
tion can be divided into three parts
W = U
∗
+ V = U + U
⊥
+ U
int
, (1.3.7)
where U is the total energy of longitudinal vibrations given by (1.2.4); U
⊥
is the
energy of transverse (bending) vibrations
U
⊥
=
α
8a
2
∑
n
η
4
n
+
1
2
β
∑
n
(η
n+1
− η
n
)
2
, (1.3.8)
and U
int
is the energy of the interaction between transverse and longitudinal vibrations
U
int
=
α
2a
∑
n
ξ
n
η
2
n
. (1.3.9)
The presence of the energy (1.3.9) means that in a nonlinear approximation the
independence of longitudinal and transverse vibrations in a chain vanishes.
We compose the nonlinear equations of motion by using
m
d
2
u
n
dt
2
= −
∂W
∂u
n
, m
d
2
v
n
dt
2
= −
∂W
∂v
n
.
Simple calculations lead to
m
d
2
u
n
dt
2
= α (ξ
n+1
−ξ
n
)+
α
2a
η
2
n+1
−η
2
n
, (1.3.10)
m
d
2
v
n
dt
2
= α (ξ
n+1
η
n+1
− ξ
n
η
n
)+
α
2a
2
η
2
n+1
η
n+1
− η
2
n
η
n
−β (η
n+2
−3η
n+1
+ 3η
n
−η
n−1
) .
(1.3.11)
These equations allow us to describe the bending vibrations, taking into account the
influence of longitudinal chain vibrations. In order to derive the nonlinear equations
of vibrations in a continuous approximation we make u se of the expansions such as
(1.2.8) and (1.3.3), retaining the higher space derivatives and the leading nonlinear
terms containing the functions u(x) and v (x):
∂
2
u
∂t
2
= s
2
∂
2
u
∂x
2
+
1
2
s
2
∂
∂x
∂v
∂x
2
; (1.3.12)
∂
2
v
∂t
2
= s
2
∂
∂x
∂u
∂x
∂v
∂x
+
1
2
s
2
∂
∂x
∂v
∂x
2
∂v
∂x
− a
2
A
2
∂
4
v
∂x
4
. (1.3.13)
The total potential energy of a linear chain (1.3.7), in the long-wave approximation
corresponding to (1.3.12), (1.3.13), is equal to
W =
1
2
a
2
α
∂u
∂x
2
+ α
∂u
∂x
∂v
∂x
2
+
α
4
∂v
∂x
2
∂v
∂x
2
+ a
2
β
∂
2
v
∂x
2
2
dx
a
. (1.3.14)
1.4 Solitons of Bending Vibrations of a Linear Chain
33
Using the rules of functional differentiation it is easy to find from (1.3.14) the forces
acting o n vibrating atoms and leading to (1.3.12), (1.3.13).
Neglecting anharmonicities of transverse displacements reduces (1.3.12) to (1.1.14)
with the dispersion law ω = sk and simplifies (1.3.13). If the linear chain is free (static
stresses are absent), the nonlinear term retained in (1.3.13) that contains ∂u/∂x is also
small. If the action of external forces generates a considerable static homogeneous
deformation ∂u/∂x =
0
= constant, (1.3.13) in a linearized form is reduced to
v
tt
=
0
sv
xx
− a
2
A
2
v
xxxx
. (1.3.15)
Equation (1.3. 15) is associated with the dispersion relation
ω
2
=
0
s
2
k
2
+ a
2
A
2
k
4
. (1.3.16)
It is interesting that the relation (1.3.16) is similar to the dispersion law (1.2.13) but
with the opposite sign in front of the term proportional to k
4
.
For ak
0
(s/A ) the dependence typical for the acoustic branch follows from
(1.3.16)
ω =
√
0
(sk )=s
∗
k. (1.3.17)
Its sound velocity s
∗
= s
√
0
is small. Thus, the long-wave dispersion law of
bending vibrations of a linear chain that experienced a static longitudinal stretching
does not differ qualitatively from the dispersion law of longitudinal vibrations of this
chain.
In the region of wavelengths for which
√
0
(s/A ) ak 1, we obtain the
dispersion law
ω = aAk
2
,
which is typical for bending waves.
1.4
Solitons of Bending Vibrations of a Linear Chain
We analyze (1.3.12), (1.3.13) to clarify their purely nonlinear properties. Recall that
the interest in the above equations is explained by the fact that they model the dynam-
ics of homopolymer molecules.
To simplify the system of nonlinear equations (1.3.12), (1.3.13), we introduce, in-
stead of time, a variable τ = st. The one-dimensional parameter aA/s will then
remain in (1.3.12), (1.3.13). Estim ating it we assume that with regard to order of
magnitude, A ∼ s. The equa tions will be written in the form
u
ττ
=
∂
∂x
u
x
+
1
2
v
2
x
; (1.4.1)
v
ττ
=
∂
∂x
u
x
+
1
2
v
2
x
v
x
−
aA
s
2
v
xxx
. (1.4.2)
34
1 Mechanics of a One-Dimensional Crystal
We remind ourselves of the notations u
x
= ∂u/∂x, v
x
= ∂v/∂x, v
xx
= ∂
2
v/∂x
2
,
etc. It is easy to find the solutions to (1.4.1), (1.4.2) in the form of a stationary profile
whose dependence on the coordinate and time is represented through a combination
ζ = x −Vτ. The waves of a stationary profile are the solutions to a system of ordinary
differential equations
(1 − V
2
)u
xx
+
1
2
d
dx
v
2
x
= 0, (1.4.3)
V
2
v
xx
+
aA
s
2
v
xxxx
=
d
dx
u
x
+
1
2
v
2
x
v
x
. (1.4.4)
The equations given admit one trivial integration. To perform this, we assume the
linear chain experiences a static longitudinal stretching (longitudinal strain) equal to ε
0
(ε
0
1). Besides that, as we are interested pr imarily in the solitary waves, we assume
all velocities and all gradients vanish at infinity. Under such boundary conditions we
get from (1.4.3)
u
x
= ε
0
−
v
2
x
2(1 − v
2
)
; ε
0
= constant. (1.4.5)
It follows then from (1.4.4) that
(V
2
−ε
0
)v
x
+
aA
s
2
v
xxx
=
V
2
2
v
2
x
v
x
V
2
−1
. (1.4.6)
We use the fact that (1.4.6) involves only the derivatives of the vector function v
and denote w = v
x
. Equatio n (1 . 4.6) will then take the form
w
xx
+(γ + βw
2
)w = 0, (1.4.7)
where
β =
1
2
s
aA
V
2
1 −V
2
, γ =
s
aA
V
2
− ε
0
.
We introduce the amplitude and the phase ϕ of the transverse motion velocity by
means of the relation
w = w(i
1
cos ϕ + i
2
sin ϕ),
where i
1
, i
2
are the unit vectors of two coordinate axes perpendicular to the direction
of a nondeformed chain. The vector equation (1.4.7) will then be reduced to the two
equations
w
xx
− wϕ
2
x
+ βw
3
+ γw = 0; (1.4.8)
d
dx
w
2
ϕ
x
= 0. (1.4.9)
Equation (1.4.9) has the form of the area conservation law (if by the variable x we
understand the time) and gives the integral of motion
I = w
2
ϕ
x
= const. (1.4.10)