Marshall L. Stoller, Maxwell V. Meng-Urinary Stone Disease
Подождите немного. Документ загружается.


328 Shekarriz
first review the epidemiology and pathophysiology of cystinuria followed by a discus-
sion on genetic aspects of the disease. Unique clinical aspects of this disorder and
management issues are presented.
HISTORY
It is interesting that the first cystine calculus was discovered in the bladder by
Wollaston in 1810 (2). He called it “cystic oxide” referring to the origin of the stone from
the bladder (kystis in Greek) (3). Berzellius (4) recognized that the compound was not
an oxide, but he also thought that the material originated from the bladder and he named
it cystine. The definitive chemical structure was described by Friedman (5) in 1902.
Garrod (6) referred to cystinuria as an inborn error of metabolism of sulfur-containing
amino acids in his lecture in 1908. This erroneous belief continued until the middle of
the 20th century. Subsequent studies by Dent and Rose (7) demonstrated that excretion
of dibasic amino acids is abnormally high in cystinuria while the serum levels of these
amino acids are normal. Harris and colleagues (8) described the genetic basis of cysti-
nuria, including its autosomal recessive inheritance. Crawhall et al. (9) described the use
of penicillamine as drug therapy for cystinuria in 1963.
Our understanding of the pathogenesis of this disease has improved by the recent
molecular developments triggered by discovery of a transporter molecule named rBAT
and the localization of the gene to chromosome 2p (10,11). Mutation in the rBAT gene
was found to be responsible for type I cystinuria and was first reported in 1994, initi-
ating discovery of multiple additional mutations for both type I and non-type I cysti-
nuria (10).
EPIDEMIOLOGY
Cystinuria has a wide range of incidence worldwide. Newborn screening programs
have estimated the disease frequency at 1:2000 in England, 1:4000 in Australia, 1:1900
in Spain, and 1:15000 in the United States. However, this frequency may be an over-
estimation due to the inclusion of non-type I heterozygotes (11). A much higher fre-
quency of the disease has been noted in Libyan Jews living in Israel, with an estimated
frequency of 1:2500. In a school screening student program, 4% of Libyan Jewish
students had heterozygote levels of urinary cystine (12). Caucasians are affected more
frequently; men and women are affected equally (13).
Cystine stones account for 2% of urinary calculi in adult patients and 6–8% in children
(14). The peak age of onset of urinary stone disease is during the third decade of life. The
only known clinical presentation of cystinuria is urolithiasis.
RENAL AND INTESTINAL TRANSPORT
OF CYSTINE AND DIBASIC AMINO ACIDS
Under normal circumstances, amino acids are freely filtered at the glomerulus and
reabsorbed almost completely at the proximal convoluted tubule. Some amino acids
such as
L-histidine and L-glycine have a higher fractional excretion at 6% and 3.5%,
respectively. Fractional excretion of most amino acids is less than 1%, resulting in a low
urinary concentration of amino acids (15). The fractional excretion of cystine is 0.4%.
Microperfusion studies have demonstrated that the proximal convoluted tubule is the
primary site for amino acid reabsorption (17).

Chapter 18 / Cystine Stone Disease 329
The reabsorption of amino acids, including cystine in the proximal tubule, involves
active and passive processes. Amino acids are reabsorbed from the tubular lumen into
the proximal cell via the brush border membrane and exit the cell via the basolateral
membrane (16). Several protein transporters mediate amino acid movement to transport
across the mammalian cell membrane. These transporters recognize and bind amino
acids and shuttle them between the intracellular and extracellular compartments.
For most organic solutes filtered through the glomerulus, including cystine, two renal
transport systems have been described (17). It is thought that 80–90% of the filtered load
is reabsorbed by a high-capacity, low-affinity system, while the remaining 10–20% is
extracted by a high-affinity lower capacity system. The high affinity system is shared
between dibasic amino acids (18). Functional studies have demonstrated that the low-
affinity transport is located in the proximal convoluted tubules (S1 and S2 segments)
whereas the high-affinity transport system is located in the proximal straight tubule (S3
segment) (19). The high affinity transporter has a low maximum rate of transport and is
competitively inhibited by other dibasic amino acids. The low affinity transporter has a
high maximum capacity and is only inhibited by arginine. Furthermore, only the high
affinity transporter is sodium independent. These two transport systems participate
equally in the reabsorption of cystine under normal filtered cystine loads. Classic cysti-
nuria (type I) results from a defect in the high affinity transport of cystine through
epithelial cells of the intestinal tract and renal tubules, which is also shared by cationic
amino acids (20).
In the small intestine, amino acids and oligopeptides are end products of protein
ingestion after digestion by pancreatic enzymes. There are two specific transport mecha-
nisms responsible for the transepithelial transport of amino acids. Amino acids are
carried by the specific transport mechanism located at the intestinal brush border of
intestinal epithelial cells. Another transport system then translocates the amino acids
across the basolateral membrane into the blood. In vitro studies of mucosal biopsies
from cystinuric patients have demonstrated that only a single intestinal transport system
for cystine exists (21).
MOLECULAR AND GENETIC BASIS OF CYSTINURIA
Cystinuria is characterized by defects in the renal and intestinal transport of dibasic
amino acids (cystine, ornithine,lysine, and arginine). In the proximal tubule, the high-
affinity transporter transports all the dibasic amino acids. Therefore, this system has
been postulated to be involved in classic cystinuria.
Traditionally, cystinuria has been classified into three types based on the observation
of the intestinal mucosal biopsies in homozygotes and urinary cystine excretion in
heterozygotes (22). All homozygotes demonstrate high levels of aminoaciduria. Type
I heterozygotes show normal aminoaciduria whereas types II and III heterozygotes
show high and moderate levels of cystine and other dibasic amino acid excretion, respec-
tively. The oral cystine load test results in an increase in plasma cystine concentration
in type III, but no change in types I and II. The intestinal active transport of cystine,
arginine, lysine or ornithine is absent in homozygotes type I and II, while it is normal
in type III [Table 1]. The proposed defect in renal and intestinal transport of cystine and
other dibasic amino acids was initially believed to be related to a single gene encoding
for both transport systems (23). Different phenotypic expressions in the intestinal trans-
port of cystine and dibasic amino acids in cystinurics were therefore explained by allelic

330 Shekarriz
mutations of the same gene (24). However, early studies demonstrated that type I cysti-
nuria is inherited in an autosomal recessive manner whereas types II and III are inherited
in an incompletely recessive manner, indicating different genes involved for type I and
the other two types (23).
The traditional classification does not differentiate between the sub-types based on
the clinical symptoms and presentation. Type I heterozygous patients have normal uri-
nary excretion of cystine and remain asymptomatic. In contrast, some type II and III
heterozygotes present with symptoms indistinguishable from the homozygotes with
significant overlap in urinary cystine excretion (13). Most homozygotes are symptom-
atic, but some may never develop stones (Table 1).
This classification correlates poorly with current molecular data and has been modi-
fied as type I and non-type I (divided clinically as type II and III) cystinuria (25). Type
I and nontype I cystinuria differ clinically on the basis of the urinary cystine and
dibasic amino acid concentrations in heterozygotes. While the type I heterozygotes are
clinically silent with normal urinary cystine levels, the non-type I heterozygotes may
present with a wide range of elevated urinary cystine levels. Abnormal urinary cystine
excretion is not always associated with clinical stone disease. The term “acalculus
cystinuria” describe a group of patients with abnormal urinary cystine and no evidence
of urolithiasis (26).
Table 1
Classification of Cystinuria: Phenotypic–Genotypic Correlations
Harris et al. (8) Completely recessive Incompletely recessive
Rosenberg et al. (24) Type I Type II Type III
Molecular Type I Non-Type I
• Urinary cystine (heterozygotes) Normal Highly elevated Elevated
(10-fold) (2-fold)
• Plasma cystine (oral load test) No change No change Increase
• Intestinal transport (homozygotes) Absent Reduced Reduced
• Protein rBAT b
0,+
AT (BAT1)
• Amino acid transport system b
0,+
b
0,+
Heteromeric rBAT/b
0,+
b
0,+
Heteromeric rBAT/b
0,+
AT
• Localization in proximal tubule S3 > S1-S2 Pars recta S1-S2 > S3 Pars convoluta
Transporter characteristic High affinity, low capacity
a
Low affinity, high capacity
b
• Gene involved SLC3A1 SLC7A9
• Chromosome 2p16.3-21 19q13.1
• No. of mutations >60 35
• Clinical symptoms
B Homozygotes Symptomatic Most symptomatic (90%)
B Heterozygotes Asymptomatic Few sy
mptomatic (13%)
a
High affinity, low capacity transporter accounts for 10% of total cystine reabsorption.
b
Low affinity, high capacity transporter accounts for 90% of total cystine reabsorption.
Data from References 13, 26, 27.
Chapter 18 / Cystine Stone Disease 331
During the last two decades, several mammalian cell membrane transport systems
have been identified. The identification and cloning of a type II membrane glycoprotein,
rBAT, was a breakthrough in our understanding of molecular basis of cystinuria. rBAT
was associated with a b
0,+
-like activity of membrane transport (22,29). The membrane
transport system b
0,+
was first described in mouse blastocytes and is a high affinity,
sodium independent transport system for cationic and zwitterionic (neutral) amino acids
(30). It was demonstrated that rBAT induces a sodium independent b
0,+
-like membrane
transporter activity in Xenopus oocytes with transport of cystine and other dibasic
aminoacids (10). When rBAT was first identified, analysis of its amino acid structure
suggested a type II membrane glycoprotein with a cytoplasmic N-terminus and extracel-
lular carboxyl (C) terminus; two to four membrane-spanning domains and a bulky cy-
toplasmic tail were described (31). However, further studies revealed that it has only
a single transmembrane domain (26,29). The structure of rBAT does not make it a
suitable candidate for a transport molecule, with the expected multimem-
brane-spanning transport protein, which would result in a suitable polar environment for
amino acid passage through the plasma membrane. Furthermore, rBAT did not transport
amino acids when induced in COS-7 cells. Thus, it was apparent that rBAT served as a
transport activator or co-transporter and there were other molecules involved in the
transport system for cystine.
Fig. 1. Plain radiographs demonstrating large cystine stone with complete staghorn configu-
ration.

332 Shekarriz
The discovery of a family of heteromeric amino acid transporters from the plasma
membrane of mammaliam cells has given us insight into the molecular events involved
in cystinuria (27). Heteromeric amino acid transporters are comprised of two subunits,
a heavy subunit (rBAT and 4F2hc) and a corresponding light subunit (LAT-1, LAT-2,
asc-1, y
+
LAT-1,y
+
LAT-2, xCT, and b
0,+
AT) linked by a disulfide bridge (27). Sequence
analysis of the rBAT gene shows it to be 30% homologous to 4F2hc. Co-expression of
these heavy and light subunits results in various amino acid transport systems. For
instance, co-expression of 4F2hc and y
+
LAT-1 induces system y
+
L amino acid transport,
which is responsible for the sodium-independent efflux of dibasic amino acids from
cells, whereas co-expression of rBAT and b
0,+
amino acid transport, which is responsible
for the renal reabsorption of cystine and dibasic amino acids at the brush border of
epithelial cells (29). In the kidney and intestine, it is postulated that rBAT forms
heterodimers through disulfide linkages to the (b
0,+
AT) to a light subunit of a 40–50 kDa
protein allowing the proper configuration to act as a transporter (32).
Lysine intolerance and cystinuria are examples of recessive disorders associated
with mutations of these heteromeric transport systems. As mentioned, 4F2hc/y
+
LAT-
1 complex accounts for the y
+
L system (catonic amino acid transport system at the
basolateral surface of intestinal and renal proximal tubular cells). Mutations of y
+
LAT-
1 gene (SLC7A7) on chromosome 14q11-13 cause the recessive disease (27). Simi-
larly, mutations in rBAT(SLC3A1) or b
0,+
AT (SLC7A9) genes will result in alteration
of the b
0,+
amino acid transport (responsible for the renal reabsorption of cystine and
dibasic amino acids at the brush border membrane) causing genetically distinct types
of cystinuria.
Type I Cystinuria
The b
0,+
system type activity associated with rBAT expression in the brush border of
the renal and intestinal epithelial cells made this molecule a candidate for the cystinuria
gene. In 1992, the first gene involved in cystine and dibasic amino acid transport was
cloned in the rabbit, rat and later in humans (10,33–35). Using an expressional cloning
approach, a 2.3 kilobase complementary DNA segment was isolated. When expressed
in Xenopus oocytes, this DNA segment induced sodium independent transport of cystine
and other dibasic amino acids. The clones isolated from rat kidneys were referred to as
NAA-Tr and D2, while that isolated from rabbit kidney cortex were referred to as rBAT.
Subsequently, the human cDNA, also 2.3 kilobases, was isolated and sequenced using
the same technique. The human cDNA, referred to as D2H and rBAT, also induced
dibasic amino acid transport when expressed in Xenopus oocytes (10,34). The genome
data base nomenclature committee designated this gene solute carrier family 3, member
1, which is abbreviated as SLC3A1 (36). The human rBAT homologue is a 45-kb, ten
exon human gene, transcribed into a 2.3 kilobases mRNA, encoding a 685 amino acid
glycoprotein with one putative membrane-spanning domain. The SLC3A1 (rBAT) gene
was mapped to the short arm of human chromosome 2 (2p21) (11). Calonge et al. (12)
reported the first rBAT mutation and concluded that it was associated with type I cysti-
nuria. At present, over 60 distinct mutations including nonsense, missense, splice site,
frameshift, and large deletions have been reported in the rBAT gene of patients with type
I cystinuria (27). It has been demonstrated that these mutations inhibit the transport of
cystine and dibasic amino acids in Xenopus oocytes. All known rBAT mutations, whether
missense or large gene deletions, are thought to cause loss of transport function in some
fashion and are fully recessive. Thus, family members who are heterozygous for rBAT

Chapter 18 / Cystine Stone Disease 333
mutation excrete cystine in the normal range (<100 mg/24 h), while affected patients
may excrete 350 mg/24 h.
The rBAT gene product is similar to the 4F2 heavy chain (4F2hc) that is involved in
other amino acid transport. It is believed that rBAT and 4F2hc are not transporters, but
rather they represent specific guidance and activator molecules for selected amino acid
transporters.
The majority of amino acid reabsorption (80–90%) occurs in the pars convoluta (S1,
S2). However, rBAT is mainly expressed in the parts recta (S3) of the proximal tubule
(Table 1). Patients who are homozygous for rBAT mutations may excrete up to 100%
of the filtered cystine load. One explanation for this finding may be the contribution of
tubular back-leak along the S3 segment. It has been postulated that patients with two
defective rBAT genes are unable to handle this back leak in addition to the portion of the
filtered load that escapes the convoluted segment (37). In contrast, rBAT heterozygotes
have adequate transport activity to ensure normal excretion of cystine and dibasic amino
acids.
Non-Type I Cystinuria
It has been long recognized that there is a wide range of phenotypic differences among
patients presenting with cystinuria. Linkage analysis and genotype-phenotype correla-
tion data have demonstrated the presence of genetic heterogeneity in cystinuria. Calonge
and associates (38) performed linkage analysis with the rBAT gene in families with type
I or type III cystinuria or both, and found that only type I cystinuria was attributable to
mutations in the rBAT gene, whereas other loci could be responsible for types II and III
cystinuria. Similarly, using linkage analysis, Biceglia and colleagues (39) reported that
type III cystinuria locus was on chromosome 19(19q13) between D19S414 and D19S220.
In 1999, the SLC7A9 (BAT1) gene was isolated. The gene encodes a 487-amino acid
protein and was mapped to chromosome 19 (19q13) with a location identical to that
predicted by linkage analysis.
Mutations in the SLC7A9 gene cause nontype I cystinuria. Data has demonstrated
several mutations of this gene in associaton with non-type I cystinuria. Data from the
International Cystinuria Consortium demonstrated 35 mutations accounting for 79% of
the carrier chromosomes in 61 non-type I patients (40). The G105R was the main non-
type I cystinuria allele (25%). Missense mutation G105R, V170M and R33W resulted
in a severe phenotype in heterozygotes with complete loss of transport activity. Other
mutations such as A182T demonstrated the lowest excretion rate of cystine and dibasic
amino acids in heterozygotes. These data collectively provide an explanation for the
phenotype variability among heterozygotes and it has been shown that mutations affect-
ing small side chains (glycine, serine, or alanine) in the transmembrane domains of b
0,+
AT are associated with severe phenotype in heterozygotes.
The SLC7A9 product is a membrane protein with 12 membrane-spanning regions.
This protein was termed b
0,+
AT for b
0,+
amino acid transporter. The dominant hypoth-
esis is the rBAT and b
0,+
AT are subsunits of b
0,+
amino acid transporter. As noted earlier,
Palacin et al. (41) postulated that these heterodimers constitute the functional units of the
b
0,+
transporter in renal and intestinal brush border membranes. The membrane spanning
protein encoded by SLC7A9 conforms to the properties of a typical transport channel
and strongly supports this hypothesis. A recent study investigated the localization and
functional properties of human cystinuria related transporter hBAT1/b
0,+
AT (42). The
cDNA encoding hBAT1 was isolated from human kidney and mapped to the human

334 Shekarriz
chromosome locus 19q12-13.1 using in situ hybridization. Tissue distribution and
expression was analyzed using northern blot and immunohistochemistry. They found
that hBAT1 message was predominantly expressed in the kidney and the protein was
localized to the apical membrane of the proximal tubule. Similar to previous data from
rat and mouse, hBAT1 when co-expressed with a type II glycoprotein rBAT in COS-7
cells exhibited transport activity with the characteristic of the amino acid transport
system b
0,+
, which transports cystine as well as basic and neutral amino acids in a
sodium-independent manner.
Although this data supports that rBAT and b
0,+
AT are subunits of the b
0,+
amino acid
transporter, this hypothesis is challenged by the different local expression of these
molecules in the proximal renal tubules. The rBAT protein is located predominantly in
the brush border membrane of pars recta (S3 segment) of proximal tubule with a decreas-
ing gradient toward the pars convoluta (S1 segment) while the opposite is true for b
0,+
AT,
which has the maximal expression in S1 segment of the proximal convoluted tubule
(Table 1) (32,43). This may explain the differences between the severity of clinical
presentation in patients with heterozygosity for rBAT or b
0,+
AT mutations. High expres-
sion of b
0,+
AT in the pars convoluta (S3) corresponds to high capacity, low affinity
transport system responsible for 90% of cystine reabsorption (21). This would explain
cystinuria in heterozygotes with b
0,+
AT mutations.
Furthermore, the partial co-localization of these proteins indicates that additional
light subunit heterodimers for rBAT or heavy subunit heterodimers for b
0,+
AT may exist
(44). Further investigation and identification of these proteins may provide us with a
better understanding of the molecular basis of cystinuria and the differences between
SLC3A1 and SLC7A9 mutations.
In our laboratories, we have recently developed a murine knockout model for type I
cystinuria. The targeted mutation of rBAT was based on a severely affected patient. This
patient and his father were genotyped by sequencing each exon and were found to have
two mutations in exon 10 that caused the missense mutations M618L and R663K. The
murine rBAT (counterpart of the human SLC3A1) gene was isolated from a Sal I mouse
genomic DNA library screen. Targeted deletions in this gene eliminated cystine trans-
port. This construct was used to produce mice in whom the endogenous SLC3A1 gene
had this truncation. The hypothesis is that mice with a targeted deletion of rBAT will
serve as an appropriate model of cystine nephrolithiasis and will exhibit features of
urinary stone disease. The preliminary data demonstrate a stone-forming clone.
This model provides us with the opportunity to perform biochemical, pathological and
molecular studies to investigate multiple steps involved in cystine stone formation.
Furthermore, medical inhibition of cystine crystallization using inhibitors can be per-
formed in vivo. Finally, this model allows us to investigate the effect of various medical
interventions on stone formation in vivo to develop more effective treatment for recur-
rent stone formation.
CLINICAL PRESENTATION
Patients present with symptoms related to nephrolithiasis. The clinical presentation
is undistinguishable from the more common calcium oxalate calculi. Therefore, a fam-
ily history is an important clue in the initial diagnosis. Recurrent and multiple stones
are common in cystinuria. Furthermore, cystine calculi may develop a large burden in
a “staghorn” configuration. Traditionally, this terminology was chosen when the calcu-
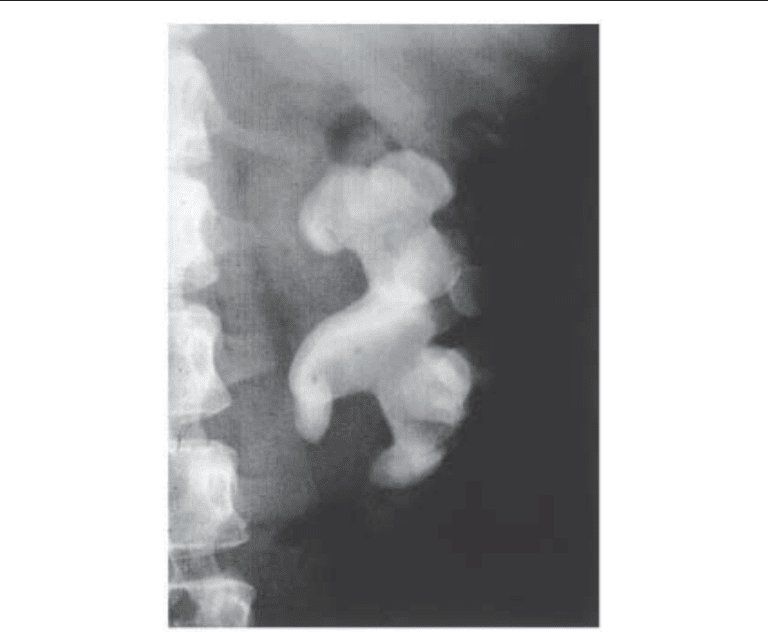
lus fills the collecting system and the shape resembles the antlers of a male elk (Fig. 2).
Chapter 18 / Cystine Stone Disease 335
By definition, staghorn calculi fill the majority of the collecting system including renal
pelvis and branches into the majority of calyces (45).
Aside from the similarities in clinical presentation of cystine and calcareous calculi,
mixed stones can account for up to half of the stones in cystinuria (46). Patients with
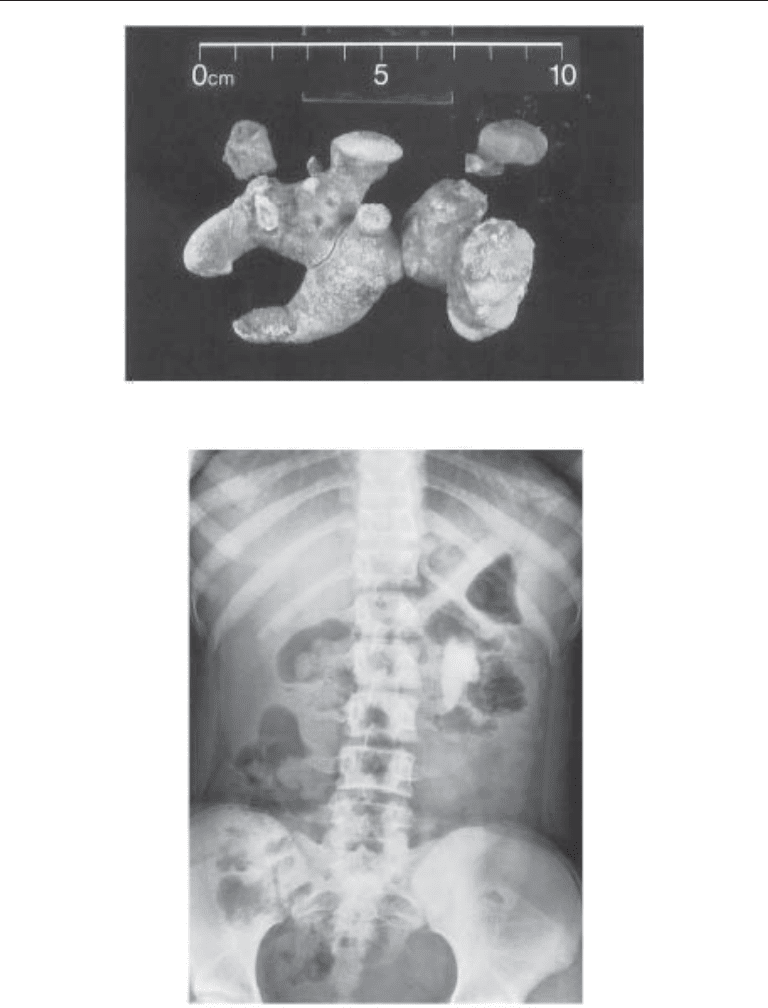
Fig. 2. Surgically removed staghorn cystine calculus.
Fig. 3. Plain radiograph of a large partial cystine stone in a 12-yr-old child with noncompliance
with medical management.

336 Shekarriz
homozygous cystinuria may have stones with calcium oxalate as the major and cystine
as the minor component. The presence of associated physiological defects such as
hypercalciuria (5.3–18.5%), hyperuricosuria (7–22%), and hypocitraturia in the
cystinuric population has been demonstrated (25,47). An association between cysti-
nuria and hyperuricemia and uric acid nephrolithiasis has also been reported (48).
Therefore, metabolic evaluation should be considered in all patients with cystine or
mixed calculi (47).
Other conditions associated with excessive cystine excretion such as Fanconi syn-
drome and Wilson’s disease should be considered (25). Furthermore, cystinuria has been
associated with several genetic diseases including mongolism, retinitis pigmentosa,
muscular dystrophy, hereditary pancreatitis and hemophilia (49). Infants younger than
6 mo may have increased cystine excretion due to immaturity of renal tubules (50).
Urinary tract infections have been reported in up to 34% of patients with cystine calculi
(51,52).
DIAGNOSIS
The diagnosis of cystine stones should be suspected in any patient with a history of
renal stones in childhood, recurrent episodes of nephrolithiasis, a strong family history
of stone disease, or amber colored calculi.
Normal urinary cystine excretion is less than 20 mg/d. The upper limit of solubility
of cystine under physiological urinary pH is 300 mg/L. By definition, a urinary cystine
concentration of above 150–250 mg/d is abnormal in adults. Stone forming children may
excrete lower amounts of cystine (75 mg/d). The initial step in the diagnosis of cystine
stones involves the examination of fresh urine for characteristic hexagonal cystine crys-
tals. Although pathognomonic, these crystals may be difficult to identify in dilute or
alkaline urine. Urinary acidification to a pH of 4.0 with overnight refrigeration before
centrifugation has been recommended to avoid infection or a rise in urinary pH. Never-
theless, these crystals are only seen in a minority of cystinuric patients (52).
A qualitative cystine test can be performed on fresh urine using the colormetric
cyanide nitroprusside test. A purple red color after addition of sodium nitroprusside and
sodium cyanide suggests a cystine excretion in excess of 75 mg/L. A false positive test
may occur in patients with homocystinuria and acetonuria. Furthermore, due to high
sensitivity of this test, heterozygous, asymptomatic patients may present with positive
results. The use of sulfur-containing drugs may also result in a false positive test due to
a nonspecific binding of nitroprusside complex with sulfide groups (16).
A positive qualitative test suggests the diagnosis of cystinuria. Therefore, if the nitro-
prusside test is positive, urinary cystine excretion should be quantitated. Quantitative
cystine excretion can be determined by ion-exchange chromatography. The total cystine
excretion in a 24-h urine collection determines the basis for a clinical diagnosis of
cystinuria. A urinary cystine excretion above 250 mg/g creatinine usually indicates
homozygous cystinuria. The definitive diagnosis is made by stone analysis.
On routine flat-plate radiographic examination, cystine stones are radiopaque (ground
glass appearance) owing to the density of the sulfur atoms (Fig. 3). They are less radio-
paque than calcium stones (53). However, there is significant variability in the radio-
density of cystine stones due to frequent mixed composition (16). Structurally, cystine
stones are generally homogenous without striations and are more rounded in appearance.
Ultrasonography may be used for diagnosis of renal, ureteral, and bladder cystine calculi
Chapter 18 / Cystine Stone Disease 337
(Fig. 4A–C). This modality will also provide information with regard to the presence and
degree of urinary obstruction, which may require more acute intervention. Similar to
other stone types, other imaging modalities, which can provide functional information,
include intravenous pyelogram and computerized tomography (CT). Intravenous pyelo-
gram was used commonly in the past and has been replaced by CT scan for acute
diagnosis of obstructing stones in most institutions.
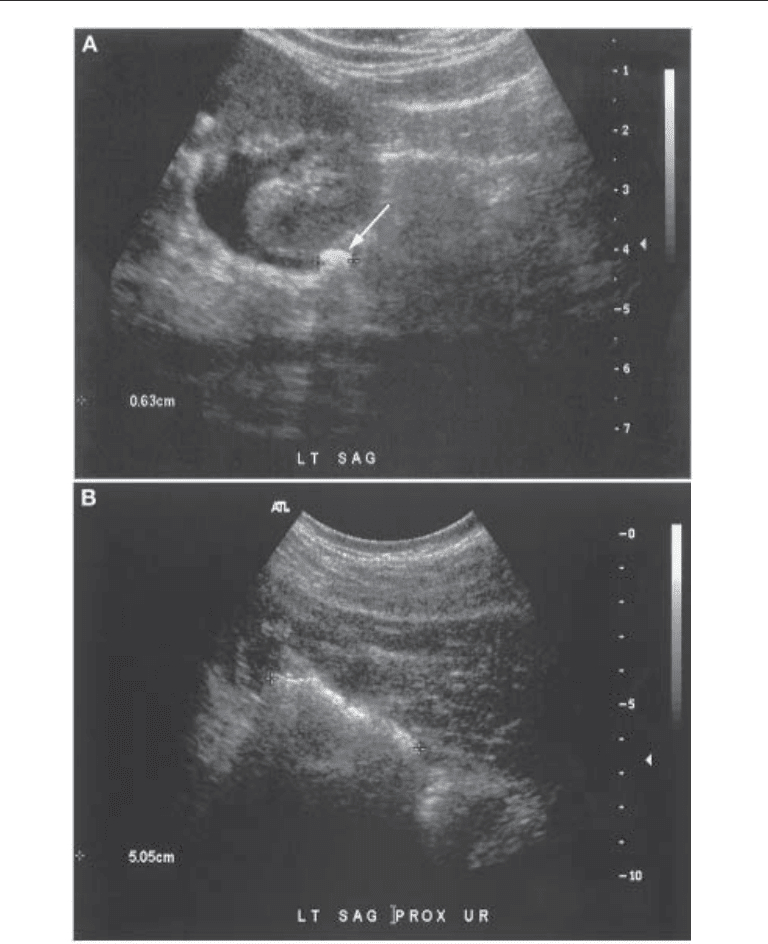
Fig. 4. Ultrasonographic diagnosis of ureteral (A), renal (B), and bladder (C) cystine calculi (see
next page). Stones are echogenic with typical posterior acoustic shadowing. Note hydronephrosis
associated with large proximal ureteral stone (A).