Schlick T. Molecular Modeling and Simulation: An Interdisciplinary Guide
Подождите немного. Документ загружается.

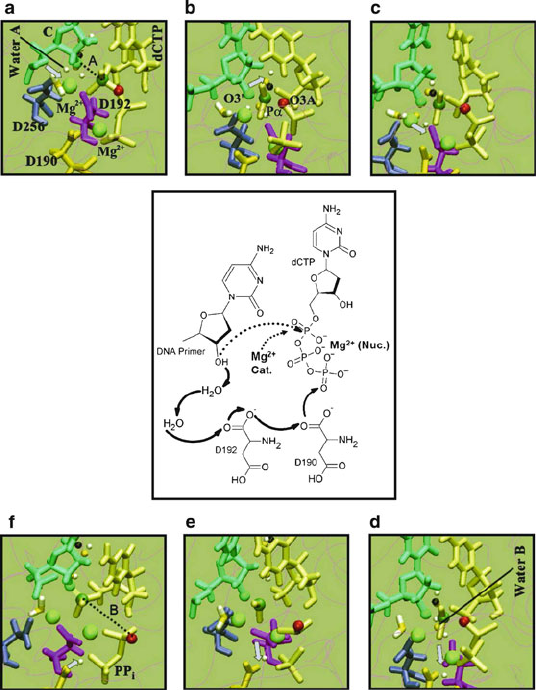
250 8. Theoretical and Computational Approaches to Biomolecular Structure
Figure 8.4. Mechanism of Grotthuss hopping chemical reaction along with key
intermediates captured during phosphoryl transfer in pol β common to the G:C and G:A
systems [1033]. Solid arrows in the scheme indicate the migration path of the proton,
and the dotted lines represent the nucleophilic attack and formation of pyrophosphate
(PP
i
)[1033]. The reaction intermediates shown are: (a) reaction state of the closed nu-
cleotide-bound enzyme state; (b) de-protonation of the the O3
H to water; (c,d) proton
transfers to Asp192; (e) proton transfers to Asp190; (f) proton reaches the pyrophosphate
unit to obtain the final product. The colors represent: blue (D256), gold (D190), magenta
(D192), yellow (dCTP), cyan (CYT: terminal DNA primer), black (the O3
H-proton),
green (the O3
oxygen, attacking nucleophile), dark green (central phosphorus), red (leav-
ing O3A oxygen), and light green (Magnesium). The oxygen and hydrogen atoms of
water molecules are in gold and white, respectively. The key distances A = O3
–P
α
and
B = P–O3A are shown; in the reaction, A decreases from 2.90
˚
A in the reactant state to
A = 1.70
˚
A in the product, and B increases from 1.7
˚
A in the reactant state to 5.8
˚
Ain
the product state. The rate limiting step in the chemical pathway is found to be the initial
de-protonation of the O3
H. The activation energy was found to be about 17 kcal/mol for
G:C and ≥ 21 kcal/mol for G:A. The QM/MM procedure involves a novel protocol using
energy minimization, dynamics simulations, and quasi-harmonic free-energy calculations.
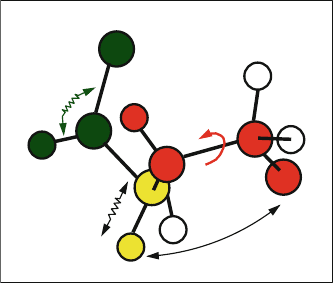
8.3. Molecular Mechanics: Underlying Principles 251
nonbonded
interaction
τ
θ
b
Figure 8.5. A molecule is considered a mechanical system in which particles are connected
by springs, and where simple physical forces dictate its structure and dynamics.
8.3 Molecular Mechanics: Underlying Principles
In theory, the overall effectiveness of molecular mechanics depends on the
validity of its three underlying principles: (1) basic thermodynamic assumption,
(2) additivity of the effective energy potentials, and (3) transferability of these
potentials.
8.3.1 The Thermodynamic Hypothesis
Does Sequence Imply Structure?
The thermodynamic hypothesis assumes that many macromolecules are driven
naturally to their folded native structure (i.e., with minimal or no intervention of
catalysts) largely on the grounds of thermodynamics. This behavior is in contrast
to other processes in the central dogma that require the crucial interplay of en-
zyme machinery to lower activation barriers. Though it is important to consider
these complex factors when modeling macromolecules, the basic thermodynamic
hypothesis of the empirical approach appears reasonable for many biomolecules
under study by molecular mechanics and dynamics.
Indeed, experiments have supported the intrinsic folding/entropy connection
for many small globular proteins [52]. Thus, a strong thermodynamic force drives
‘scrambled’ conformations of high free energy to the native state of low free
energy. Folding in many cases is reversible (denatured ⇐⇒ native state) and
attainable from many different configurations.
Theoretical analyses based on statistical studies of spin glasses further suggest
that the probability that a randomly synthesized protein will exhibit a thermody-
namically dominant fold (i.e, the global minimum of the free energy) increases
rapidly as temperature decreases [1167].

252 8. Theoretical and Computational Approaches to Biomolecular Structure
As discussed in Chapter 3, the resilience in general of overall protein structures
to small mutations — in some cases even at critical regions, as in the Rop
turn ([205] and Figure 3.10) — is another indicator of the strong thermody-
namic tendency of proteins to adopt and retain native structure/folds. For higher
organizational forms of DNA, the intrinsic topology also appears to induce a
folded configuration determined in large part by base composition and the ionic
medium [269].
The kinetics involved in such folding processes represent another subject of
intense interest (see [861,1257], the discussion in Chapter 2,andBox8.2).
Box 8.2: The Thermodynamic Hypothesis as Exemplified by The Levinthal Paradox
The concept introduced by Cyrus Levinthal in 1969 [744], which became termed the
“Levinthal paradox”, is that a protein cannot find its native state by a random search
through all its possible conformations. This is because such an exhaustive enumeration
would in theory require eons of years, while real proteins fold on the sub-second and
second timeframe. Levinthal thus hypothesized that well defined pathways must exist for
protein folding in nature [743].
Recently, protein-folding kinetics have been analyzed and interpreted on the basis of
simple simulations of polypeptide models. The “old view” that emerged from Levinthal’s
work is now being merged with the “new view” [313, 314, 707], which promotes many
competing folding pathways rather than a unique pathway with well-defined intermediates.
The two views can be merged when the folding pathways all share a unifying pattern of
native contacts and can be described within a common multidimensional energy landscape
[1385]. (See also discussion in Chapter 2).
While this protein folding problem is generally formulated purely in terms of “sequence
dictates structure”, it is clear that in many cases the folding process in vivo is enhanced
or facilitated through the external intervention of additional molecules such as chaper-
ones. Such agents may stabilize intermediates, prevent aggregates of misfolded proteins,
and/or reduce the activation energies effectively [561, 833]. Members of the chaperone
class that includes the heat-shock protein hsp70 bind to newly-synthesized proteins and
short linear peptides, possibly preventing aggregates. Other chaperones assist in protein
translocation across membranes. Members of the chaperonin class include the well-studied
GroEL/GroES complex from bacteria [387] that bind to partially-folded polypeptides and
assist in the folding. Many questions are now being investigated regarding the folding
kinetics in all these systems (see [519,1326], for example).
8.3.2 Additivity
The molecular mechanics principle of additivity assumes that the effective
molecular energy can be expressed as a sum of potentials derived from simple
physical forces: van der Waals, electrostatic, mechanical-like strains arising from
8.3. Molecular Mechanics: Underlying Principles 253
“ideal” bond length and angle deviations, and internal torsion flexibility (rotation
of two chemical groups about the bond joining them). The forces can be separated
into local (bonded) and nonbonded (nonlocal) terms.
Local Terms
The local components for macromolecules can typically be written as:
E
local
(X)=
bonds
i
E
bond
(b
i
)+
bond angles
i
E
bang
(θ
i
)+
dihedral angles
i
E
tor
(τ
i
),
(8.3)
where summations extend over the sets of bonds {b
i
}, bond angles {θ
i
},and
dihedral angles {τ
i
} (see Fig. 8.5). Functional forms are typically harmonic (e.g.,
S[b −
¯
b]
2
) and trigonometric (e.g., functions of cos(θ)), as discussed in the next
chapter. Note that all internal variables are functions of interatomic distances,
r
ij
. Cross terms (like E(b, b
) or E(b, θ)) are not commonly used for proteins
and nucleic acids because the associated force constants (‘off-diagonals’ in the
force-constant matrix) usually have much smaller values than those associated
with the ‘diagonal’ terms. Such force constants, for example S
bb
for bond/bond
interactions or K
bθ
for bond/bond-angle terms, are associated with potentials of
the form S
bb
(b −
¯
b)(b
−
¯
b
) and K
bθ
(b −
¯
b)(θ −
¯
θ), respectively, which are
common in small-molecule force fields (e.g., [30]).
Nonlocal Terms
The nonlocal components can be written as:
E
nonlocal
(X)=
nonbonded pairs
(i,j),i<j
E
r
(r
ij
), (8.4)
where the functions are often rational (e.g.,
N
i
i=1
r
−m
). These energies are
usually comprised of van der Waals and Coulombic contributions between non-
bonded atom pairs. (Nearby atom pairs counted in the local energy may be omitted
to avoid double counting).
Benefits of Separability
The natural separability into bonded (local) and nonbonded (nonlocal) terms can
be exploited in the design of minimization algorithms that use function structure
to accelerate convergence (see Chapter 11). This separability is also the basis for
multiple-timestep protocols for molecular dynamics that update local and nonlo-
cal forces at different frequencies (see Chapters 13 and 14). This difference in
force updating is reasonable because the local terms generally change rapidly,
while the nonlocal terms change more slowly, with distance and time.
While the number of nonbonded terms grows quadratically O(N
2
) with the
number of atoms, the number of local terms — involving pairs, triplets, and
quadruplets of bonded atomic sequences — grows only linearly with size.

254 8. Theoretical and Computational Approaches to Biomolecular Structure
This computational complexity of the nonbonded terms is especially a burden in
simulation protocols for large systems that require updating for many iterations,
as in macromolecular dynamics. Fortunately, some work can be reduced since the
nonbonded Coulomb forces change more slowly with distance than the bonded
terms, and hence can be updated less often than the local forces. Chapter 10 is
devoted to the nonbonded forces.
Multibody Potentials
In this typical energy formulation, nonbonded interactions are “effective pair
potentials” (i.e., additive two-body forces). For electrostatic interactions, for ex-
ample, these effective pair potentials only reflect in some average sense the charge
distribution due to molecular polarizability. It is clear that many-body contribu-
tions are important for accurate reproduction of certain molecular properties. For
example, accurate account of dispersion forces for polar molecules (i.e., mole-
cules in which the charges are nonuniformly distributed), such as water, require
three-body interaction potentials:
E
ijk
(r
ij
,r
jk
,r
ik
).
These potentials can be expressed as a composite of trigonometric and rational
functions in terms of three bond lengths and three bond angles [131]. Indeed, the
importance of water as a solvent for proteins and nucleic acids was realized in the
1970s [1344] and has prompted development of ‘polarized’ water potentials [244,
1343, for example]. In practice, these models increase the number of interaction
sites per water molecule.
8.3.3 Transferability
The principle of transferability assumes that potentials can be developed to in-
corporate all experimental data for representative structures and then be applied
successfully to the prediction of large biological molecules composed of the same
chemical subgroups. This is basically a reasonable assumption since bond lengths
and bond angles tend to adopt similar values in different molecular species under
normal conditions. However, under special straining forces, such as in cycloalka-
nes, these values may vary significantly from ‘ideal’ or average values. Modeling
the structures and properties of complex aromatic or conjugated systems
3
also
requires special treatment, such as using sophisticated quantum-mechanical cal-
culations to obtain bond orders of conjugated bonds, which can be related to
bond lengths and stretching constants, and in this way reducing the problem to
molecular mechanics ([29,899,1210,1246,1341], for example).
3
Aromatic compounds are benzene-like in structure and properties. Conjugation is a structural
feature caused by the overlap of the π orbital with other orbitals in the molecule.
8.3. Molecular Mechanics: Underlying Principles 255
Functional Variations in Geometry
In small molecules, the environment-dependence of geometric trends can be
modeled by a function rather than a constant. For example, Allinger and
coworkers devised functions for bond lengths in small-molecule force fields
(MM3/MM4) to account for the electronegativity of attached substituents [1210]
and for hyperconjugation [26]. The former parameterization [1210] can, for ex-
ample, accurately model the shorter C–C bond in fluoroethane (C
2
H
5
F) relative
to ethane (C
2
H
6
), since the fluorine is electronegative; similarly, it can also ac-
count for a longer C–O bond in an alcohol where the electropositive hydrogen is
attached to an oxygen (like ethyl alcohol, CH
3
CH
2
OH), relative to an analogous
molecule in which a carbon rather than hydrogen is attached (as in dimethyl ei-
ther, C
2
H
5
OC
2
H
5
). A functional dependence for reference bond lengths used for
hyperconjugated systems [26] can account for bond-length trends in molecular
species in which bond orbitals overlap and lead to resonance, like longer C–H or
C–C bonds in carbonyl compounds (e.g., H–C–C=O ↔ H
+
+ C=C–O).
Proliferation of Atom Types
An alternative approach for incorporating the environment dependence of geo-
metric tendencies is to increase the number of ‘atom types’. Thus, atom types
reflect the molecular environment (e.g., aromatic carbon in a nitrogenous base)
and hybridization (e.g., sp
2
or sp
3
)[175,805,1359].
There are around 160 atom types, for example, in the CHARMM force field
for proteins (version 22) and nucleic acids (version 27) [415, 804–806]: 62 car-
bons, 31 hydrogens, 28 nitrogens, 18 oxygens, 4 sulfurs, 3 phosphorus atoms,
6 fluorines, one heme iron, and 7 different ions; see Table 8.1 for some examples
of these atom types and Figure 8.6 for illustrations for selected residues [805].The
proliferation of atom types in modern force fields thus attempts to improve com-
patibility with experiment. Alternatively, force fields may be restricted to certain
families of molecules such as alkanes, amides, and carboxylic acids [185,764].
I emphasize that individual functions should not be transferred from one force
field to another, since the entire potential is parameterized as a whole to reproduce
consistently experimental data.
Overall, while the transferability assumption is inherent in this empirical
science, molecular mechanics has steadily gained recognition through many im-
portant contributions. Today’s force fields are excellent for deducing structures
and properties of many molecular systems, especially for small molecules us-
ing specialized force fields. One advantage of theoretical calculations is that the
thermal motions and lattice effects that influence crystallographically-determined
structures may not be a problem when accurate force fields are used to predict
molecular structures and properties.

256 8. Theoretical and Computational Approaches to Biomolecular Structure
Table 8.1. Examples of atom types defined in CHARMM 22 and 27 [415,804–806].
Atom Symbol Atom modifier
Carbon C polar (carbonyl, peptide backbone)
CA aromatic
CC carbonyl(Asn,Asp,Gln,Glu)
CPT inter-ring in tryptophan
CP1, CP2, CP3 special tetrahedral, in proline
CT1 aliphatic sp3 in CH
CT2 aliphatic sp3 in CH
2
CT3 aliphatic sp3 in CH
3
CN1 nucleic acid carbonyl carbon
CN3 nucleic acid aromatic carbon
Oxygen O carbonyl
OC carboxylate
OH1 hydroxyl
ON6 nucleic acid deoxyribose ring oxygen
ON6B nucleic acid ribose ring oxygen
Nitrogen N proline
NH1 peptide
NH2 amide
NN1 nucleic acid amide nitrogen
NN2 nucleic acid protonated ring nitrogen
Hydrogen H polar
HA nonpolar
HP aromatic
HS thiol
HN1 nucleic acid amine proton
HN2 nucleic acid ring nitrogen proton
8.4 Molecular Mechanics: Model and Energy
Formulation
In practice, the challenge and success of molecular mechanics rely on both
effective formulation of the potential energy function and the application of
suitable search algorithms (e.g., multivariate minimization, sampling, dynamic
simulations). With the well known difficulties of large-scale minimization (see
Chapter 11) and the timestep problem in dynamics (Chapters 13 and 14), the struc-
tural outcome — and hence biological implications of any calculation — depends
on the combination of modeling and algorithmic techniques employed. Indeed,
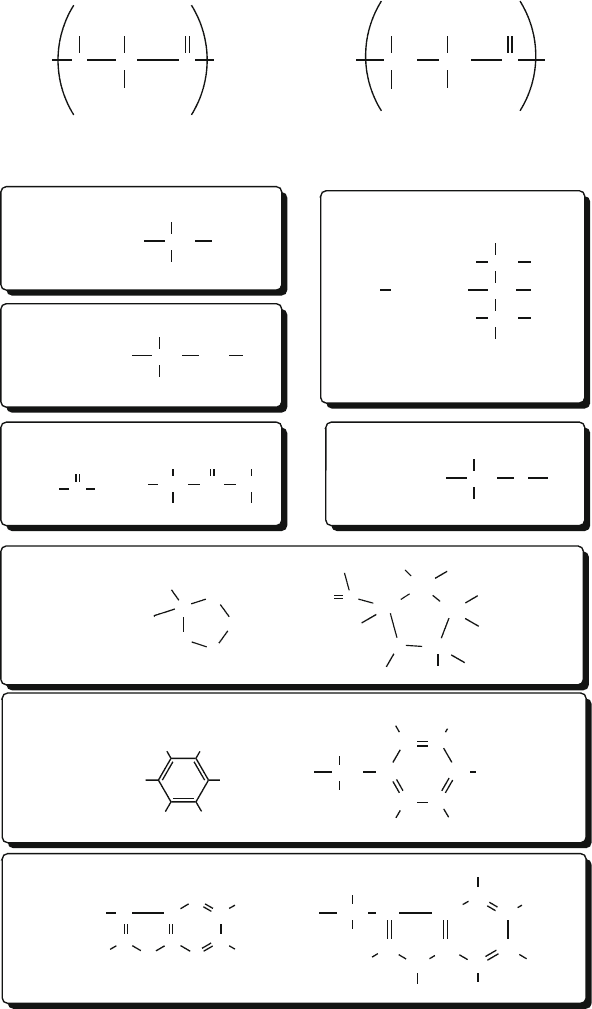
8.4. Molecular Mechanics: Model and Energy Formulation 257
C
C
N
H
C
C
C
H
C
C
H
C
C
H
2
N C
i
α
HAH
Ri
H
C
O
OH CT1
HB
Ri
C
O
NH1
H
CH (CH
3
)
2
CH
3
CT3
HA
HA
HA
CT1
CT3
HA
CT3
HA
HA
HA
HA
HA
HA
CH
2
OH
CT2
HA
HA
OH1 H
CH
2
SH
CT2
HA
HA
S HS
CH
2
C
O
NH
2
CT2
HA
HA
CC
NH2
H
H
O
CP1
N
CP3
CP2
CP2
HA
HA
C
BH
HA
HA
HA
O
HA
H
H
HH
H
CH
2
CA
CA CA
CA
CACA
HP HP
HP
HPHP
CT2
HA
HA
H
H
H
CY
CA
NY
CPT
CPT
CA
CA
CA
CA
CT2
HA
HA
HP
HP
HP
HP
H
HP
Polypeptide Building Block
Alanine R:
Serine R:
C
HN
C
H
2
CH
2
H
2
C
Proline R:
Valine R:
Phenylalanine R:
H
COO
Asparagine R:
Typical Nomenclature
Tryptophan R:
Cysteine R:
_
Figure 8.6. Examples of atom types as used in polypeptides in the CHARMM program
[805, 806].

258 8. Theoretical and Computational Approaches to Biomolecular Structure
some strengths and weaknesses of molecular mechanics have been debated openly
in a series of papers [455, 668, 1068, 1069]; see also Homework Assignment 7.
Some valid questions are:
• How accurate are quantum-mechanically derived partial charges?
• How do Cartesian and dihedral-angle representations affect results?
• Is it appropriate to introduce arbitrary scale factors in energy coefficients to
enhance agreement with experiment?
The cautions and possible pitfalls of force field derivations and parameter choices
are thus important to emphasize, even for state-of-the-art force fields.
In formulation of the energy, three basic and important decisions are involved:
(1) representative configuration space, (2) functional form, and (3) numerical
values for the parameters. We discuss each in turn.
8.4.1 Configuration Space
A Question of Size
The atomic configuration
4
space for a molecular system consists of 3N −6
degrees of freedom, where N is the number of atoms. (Six degrees of freedom
are removed for rigid-body translation and rotation invariance of the energy).
Thus, the number of degrees of freedom for proteins and nucleic acids typically
ranges in order from 10
3
to 10
4
. With explicit representation of water mole-
cules, this number rises by at least an order of magnitude (see, for example,
solvated-macromolecular system sizes in Table 1.2 of Chapter 1).
The complete set of 3N − 6 degrees of freedom can be described directly by
lists of Cartesian coordinates for all the atoms in the system. Alternatively, internal
variables such as bond lengths, bond angles, and dihedral angles may be used; this
internal representation has been quite successful for the study of proteins [1068,
for example].
Other representations have been used for biomolecules to reduce this number
of variables. Reductions are possible by fixing bond lengths and angles and work-
ing in dihedral angle space, or by restricting energetic pathways to approximate
formulations. However, these representations do not usually lead to enhanced
efficiency unless the work involved in the nonbonded computations — the real
computational ‘bottleneck’, see Chapter 10 — is reduced significantly.
4
Configuration, a more general term than conformation, is often used by mathematicians to
describe the shapes of objects in space. The more chemical term conformation often refers to
configurations that differ from one another through rotations of groups of atoms about the bond con-
necting them (i.e., dihedral angles). Note, however, that in organic chemistry a configuration refers
to stereoisomers, molecules with different connectivity arrangements which cannot be converted into
one another through rotations about bonds.
8.4. Molecular Mechanics: Model and Energy Formulation 259
The Pseudorotation Description
An example of a parameter reduction approach is the pseudorotation path
developed for nucleic acids [35,271,750]. This energetic path, initially developed
for the hydrocarbon cyclopentane [766] and later extended to ribose and deoxyri-
bose sugars, constrains the energy of five-membered sugar rings to a wavelike
motion from a mean plane. This plane can be defined in various ways by po-
sitions of the five skeletal ring atoms. See Chapter 5 for a discussion of these
descriptions in the section on furanose conformations.
While conceptually simple, the reduction in degrees of freedom from 9 (3·5−6)
to 2 is clearly approximate. The pseudorotation approximation was noted to pro-
duce anomalies in overall nucleic acid structures [937,944], inconsistencies with
ring closure, and mathematical difficulties in expressing the energy derivatives
in terms of the independent conformational variables. More generally, while for
some systems simplifications in the representative conformation space may be ac-
ceptable, it is usually advantageous to avoid constraints altogether [1101]. (The
pseudorotation concept remains a useful analysis tool, however).
Cartesian Space
Cartesian coordinate space is most convenient for direct differentiation of the
energy in terms of the independent parameters, as realized in the early days of
force field development by Lifson and Warshel [766], and therefore application
of efficient second-derivative Newton minimization methods (see Chapter 11).
Cartesian space is also most natural for implementation of molecular dynamics,
since the generated molecular trajectories
{X(t
0
),X(t
0
+Δt), ..., X(t
0
+ n Δt), ...},
where t
0
is the initial time reference and Δt is the timestep (see molecular dynam-
ics chapters), generally rely on recursive expressions. That is, the new positions
and velocities
X(t
0
+ n Δt)andV (t
0
+ n Δt)
are explicit functions of the previous positions and velocities
X(t
0
+(n −1) Δt)andV (t
0
+(n −1) Δt) .
With all these considerations, it is usually advantageous to enforce con-
straints — if necessary — by means of penalty functions (i.e., soft constraints).
Harmonic penalty functions can be used, for example, to keep bond lengths and
angles close to their observed values.
8.4.2 Functional Form
Composition
The potential energy E of a molecular model is typically constructed as the sum of
contributions from the following types of terms: bond length and bond angle strain