Schlick T. Molecular Modeling and Simulation: An Interdisciplinary Guide
Подождите немного. Документ загружается.

260 8. Theoretical and Computational Approaches to Biomolecular Structure
terms (E
bond
and E
bang
), a torsional potential (E
tor
), a Lennard-Jones potential
to model repulsion at short interatomic separations and attraction at long distances
(E
LJ
), and a Coulombic potential among the pairs of charged particles in the
system (E
coul
):
E = E
bond
+ E
bang
+ E
tor
+ E
LJ
+ E
coul
(8.5)
E
bond
=
i,j∈S
B
S
ij
r
ij
− ¯r
ij
2
E
bang
=
i,j,k∈S
BA
K
ijk
(cos θ
ijk
− cos
¯
θ
ijk
)
2
E
tor
=
ijk∈S
DA
n
Vn
ijk
2
1 ± cos(nτ
ijk
)
E
LJ
=
i,j∈S
NB
−A
ij
r
6
ij
+
B
ij
r
12
ij
E
coul
=
i,j∈S
NB
q
i
q
j
(r
ij
) r
ij
These equations represent first approximations to the physical potentials but
are nonetheless reasonable for biomolecules. In these general expressions, the
symbols S
B
, S
BA
,andS
DA
denote the sets of all bonds, bond angles, and dihe-
dral angles. The nonbonded set, S
NB
, typically includes all (i, j), i<j,atom
pairs separated by three bonds or more. Bond and angle variables capped by bar
symbols denote reference values associated with these quantities.
The 6/12 Lennard Jones potential above (i.e., attraction of form −A/r
6
and
repulsion of form B/r
12
) is typical for large-molecule force fields because of
its mathematical convenience; a Buckingham potential (with the same functional
form for attraction but an exponential repulsion term of form B exp(−B
r))[766]
is in principle closer to the electronic structure of the atom and thus provides a
better potential fit over a broader range of interparticle distances [30].
Molecular Geometry
To be more precise, we define the analytic expressions for the geometric quanti-
ties in the potential energy.
Positions and distances. For a molecular system of N atoms (possibly inclu-
ding atom groups) in Cartesian coordinate space, let
x
i
=(x
i1
,x
i2
,x
i3
),i=1,...,N,
denote the position vector of atom i,and
r
ij
≡ x
j
− x
i

8.4. Molecular Mechanics: Model and Energy Formulation 261
denote the distance vector from atom i to j. Our potential energy function E then
depends on all Cartesian variables of the atoms:
E = E(X) ≡ E(x
1
, x
2
,...,x
N
)
where X is the collective vector in the Euclidean space R
n
of dimension n =3N.
For any vector a we denote its standard Euclidean magnitude by a.For
convenience later in writing the potential energy function, we also denote an
interatomic vector magnitude r
ij
as r
ij
, in nonbold type.
Bond angles. A bond angle θ
ijk
formed by a bonded triplet of atoms i–j–k is
expressed as an inner (or dot) product:
cos θ
ijk
=
(x
k
− x
j
) • (x
i
− x
j
)
r
ij
r
kj
. (8.6)
Dihedral angles. A dihedral angle τ
ijk
, defining the rotation of bond i–j
around j–k with respect to k–l, is expressed as (see Fig. 3.14 in Chapter 3
and Fig. 8.5):
cos τ
ijk
= n
ab
• n
bc
. (8.7)
The vectors n
ab
and n
bc
denote unit normals to planes spanned by vectors {a, b}
and {b, c}, respectively, where a = r
ij
, b = r
jk
,andc = r
k
. Denoting θ
ab
and
θ
bc
as angles θ
ijk
and θ
jk
, respectively, we write:
cos τ
ijk
=
a × b
absin θ
ab
•
b × c
bcsin θ
bc
. (8.8)
The sign of τ
ijk
is set by the sign of the triple scalar product a •(b × c).
Lagrange’s identity. To simplify potential energy equations and differentiation
[1103], it is convenientto work with inner product expressions and use Lagrange’s
identity:
(a ×b) • (c ×d)=(a •c)(b •d) − (b • c)(a • d) . (8.9)
This produces the alternative expression for cos τ:
cos τ
ijk
=
(a × b) • (b × c)
[(a ×b) • (a × b)(b × c) • (b ×c)]
1/2
=
(a • b)(b • c) − (a • c)(b •b)
[(a •a)(b •b) −(a •b)
2
][(b • b)(c • c) −(b • c)
2
]
1/2
.(8.10)
According to this convention, τ =0
o
defines a cis coplanar orientation for atoms
i–j–k–l, τ = 180
o
defines a trans coplanar orientation, and a positive sign cor-
responds to a clockwise rotation of the far bond with respect to the near bond
(when viewed along the j–k bond).
262 8. Theoretical and Computational Approaches to Biomolecular Structure
Derivatives. First and second derivatives of these expressions are often needed
for structure minimization, molecular dynamics, and various conformationalanal-
yses (e.g., normal modes). The derivative expressions are tedious to derive (see
Homework Assignment 8) and care must be used to avoid singularities, but var-
ious algorithmic procedures have been developed to simplify the task in practice
[1103, for example].
8.4.3 Some Current Limitations
Parameterization details for each potential energy term, including functional vari-
ations, are described in the next chapter. We conclude this chapter by mentioning
some general limitations of current molecular force fields:
1. Many Force Field Choices. At present, there is no “universal force field”,
nor are the many force fields in use close to converging to one another
in some sense. Essentially, users around the world develop a preference
for one force field over another on the basis of their target application and
practical factors such as cost and convenience.
For example, the MM2/3/4 force-field family developed by Norman
Allinger and coworkers is a popular choice for small molecular systems
[25, 30, 696]. Protein modelers often use the CHARMM package devel-
oped by Martin Karplus and coworkers [174]. Nucleic acid modelers might
prefer the AMBER program developed by the late Peter Kollman and
coworkers [204]. Harold Scheraga’s team developed the dihedral-angle
ECEPP family of force fields for protein modeling [58, 897]. Other force
fields are also available, such as GROMOS [950], OPLS-AA [618, 629],
CFF [614], and CVFF [371]; see also [489,p.76]and[4] for lists of
available force fields and associated molecular modeling packages, and
Table 11.1 of Chapter 11 for minimization algorithm information for some
of these programs.
A current effort is the development of programs which utilize force fields
that cover a wider range of chemical systems. This, however, involves a
trade-off between force field accuracy and versatility [1123]. The MMFF
force field, for example, developed at Merck in the late 1990s [497–500],
was designed to model a wide range of organic molecules and proteins.
Still, the recent trend has been to incorporate force field segments for
specialized systems in existing modeling packages, for example to model
a complex between small organic and drug-like molecules with biomo-
lecular systems. The OPLS-AA force field, in particular, covers a wide
variety of organic molecules. Recently, general parameters for organic mol-
ecules have been developed for usage in combination with the AMBER and
CHARMM force fields through easy extensions [1300,1331].
8.4. Molecular Mechanics: Model and Energy Formulation 263
2. Variability in Functional Form and Numerical Parameters. As detailed
in the next chapter, a great deal of variability exists in the functional forms
used for each potential energy term as well as in the numerical values for
the associated parameters.
3. Inclusion of Explicit Hydrogen Bonding. Some macromolecular force
fields use explicit hydrogen-bonding potentials, though these potentials
were more common in older versions that did not consider hydrogens
explicitly. Note that the strength of a hydrogen bond is determined by
its geometry, which depends on the distances associated with the Donor-
Hydrogen ··· Acceptor sequence and the Donor-Hydrogen ···Acceptor
angle (θ
dha
) formed about the hydrogen atom. For a linear hydrogen bond
(usually strongest), θ
dha
has the ideal value of 180
o
. In the current bio-
molecular AMBER and CHARMM force fields, for example, the proper
dependence of hydrogen bonding on distance and angular orientations is
adequately treated with the electrostatic and Lennard-Jones terms.
In theory, classical electrostatic and (quantum) bonding forces can account
for hydrogen-bonding interactions. Thus much work has gone into for-
mulating appropriate hydrogen-bond potentials for small-molecule force
fields.
Hydrogen-bond potentials can be derived from crystal packing studies or
quantum-mechanical calculations, or introduced to correct weak interac-
tions [898]. A re-optimization of hydrogen-bond potentials for MM3 [768]
based on ab initio calculations concluded that hydrogen-bond interactions
are more complex than previously thought. In that work, the hydrogen
bond potential was formulated as a complex function of the Hydrogen ···
Acceptor distance, the Donor-Hydrogen bond length, and the cosine of the
θ
dha
angle. While this [768] and earlier works treated the overall nuclear
charges, more recent work has shown that a better representation involves
placing the center of the electron density where the lone pairs are and not
where the nucleus is [796].
4. Electrostatic Approximations. In the simple Coulomb potential described
above, interactions between pairs of atoms often consider only point
charges for each atom. However, the charge distribution about each nucleus
is clearly more complex, and in some cases induced dipole effects (i.e., dis-
tortion of the electron distribution) are important to consider. This notion of
modeling electronic polarizability (roughly a measure of the distortion of
an electronic charge cloud) is a continuing focus in force field design (see
[223,243,501,630,970,1335,1343] for example).
5. Limited Use of Quantum-Mechanical Information. In theory, quantum-
mechanical theory from molecular orbital techniques can be applied to
small molecules to improve functional form and assign more accurate
264 8. Theoretical and Computational Approaches to Biomolecular Structure
parameters (e.g., to fit parameters to ab initio relative energies or to the
quantum-mechanical energy surface curvature). In practice, this fitting is
not easy to perform and depends on the quality of the quantum calculations
(e.g., basis set). Yet, quantum-based calculations have been used to assign
the electrostatic partial charges, and the newer generation of force fields re-
lies on quantum calculations wherever possible (e.g., [497, 630, 768, 805,
842]), in combination with increasingly accurate experimental measure-
ments. Thus, using both experimental and quantum calculations — despite
each technique’s limitations — can provide the best overall results.
9
Force Fields
Chapter 9 Notation
S
YMBOL DEFINITION
Scalars & Functions
c speed of light
c
s
ionic concentration
e
electron charge
Planck’s constant over 2π
k
harmonic spring constant
m
particle mass
m
e
electron rest mass
n
periodicity of rotational barrier (torsional potential)
q
i
Coulomb partial charge of atom i
r
bond length
¯r
reference bond length
r
ij
interatomic distance (between atoms i and j)
x
displacement
A
ij
,B
ij
Lennard-Jones coefficients for atom pair i, j (attraction,
repulsion)
D
Morse well depth parameter
E
potential energy
E
coul
Coulomb energy
E
LJ
Lennard-Jones energy
E
r
bond length energy
E
rr
stretch/stretch energy
E
rθ
stretch/bend energy
E
rθr
stretch/bend/stretch energy
E
θ
bond angle energy
E
θθ
bend/bend energy
T. Schlick, Molecular Modeling and Simulation: An Interdisciplinary Guide, 265
Interdisciplinary Applied Mathematics 21, DOI 10.1007/978-1-4419-6351-2
9,
c
Springer Science+Business Media, LLC 2010
266 9. Force Fields
Chapter 9 Notation Table (continued)
S
YMBOL DEFINITION
E
ρ
Urey-Bradley energy
E
τ
dihedral (torsional) angle energy
E
τθθ
torsion/bend/bend energy
E
τθ
torsion/bend energy
E
χ
improper torsion energy
E
χχ
improper/improper torsion energy
F
force
F
coul
Coulomb force
K
coul
Coulomb potential constant
K
h
harmonic bending constant
K
t
trigonometric bending constant
N
number of atoms
N
e
number of outer shell electrons
S
h
harmonic stretching constant
S
m
Morse stretching constant (well width parameter)
S
q
stretching force constant for special quartic potential
V
ij
,r
0
ij
Lennard-Jones coefficients for atom pair i, j (energy
minimum/interaction distance)
V
n
barrier height of torsional potential associated with
periodicity n
α
atomic polarizability
dielectric constant
0
permittivity of vacuum
θ
bond angle
¯
θ
reference bond angle
θ
tet
tetrahedral bond angle, 109.47
o
,orcos
−1
(−1/3)
κ
Coulomb screening parameter
λ
wave number (wavelength of absorption)
μ
reduced mass
ν
characteristic frequency
τ
torsion (or dihedral) angle
τ
0
reference torsion (or dihedral) angle
χ
Wilson angle (for improper torsion potential)
ω
angular frequency
The purpose of models is not to fit the data but to sharpen the
questions.
Samuel Karlin, 1983 (1923–2007).
9.1 Formulation of the Model and Energy
In this chapter, we discuss only basic functional expressions of the potential en-
ergy function, emphasizing the simple forms typically used for biomolecules. For
biomolecular systems, computational speed is premium, and the use of more
9.2. Normal Modes 267
complex terms (higher-order expansions, cross terms, etc.), as employed for
accurate modeling of smaller systems, is not practical. The next chapter discusses
important topics related to this computational complexity of the nonbonded terms:
spherical cutoff techniques, fast electrostatic evaluation techniques (Ewald and
fast multipoles), and implicit solvation alternatives.
While improvement of potential energy functions — both in terms of func-
tional form and parameters — has been an ongoing enterprise, the current,
“second-generation” molecular mechanics and dynamics force fields are more
sophisticated than those originating from pioneering works in the 1960s and
1970s. Specifically, parameterization depends quite significantly now on quan-
tum mechanical calculations. “Third generation” force fields that account more
accurately for electronic polarizabilities are already emerging (see [223,501,630]
for example).
Parameterization of force fields ensures that calculations produce appropriate
molecular geometries and interaction energies for a set of model compounds that
are appropriate for the force field. For proteins, test compounds are peptides or
model peptides [805]. For nucleic acids, deoxyribonucleosides [218, 265]and
compounds containing the furanose ring and oligonucleotide crystals [415]are
appropriate.
Force field parameters are optimized in a ‘self consistent’ fashion [185, 766,
1152] so as to reproduce the increasing body of experimental information on
molecular geometries (from crystal and solution studies) to many other proper-
ties: measurements of vibrational frequencies, heats of formation, intermolecular
energies and geometries, torsional barriers, and more. The use of dynamic sim-
ulations to assess the quality of the force field and to refine parameters — so
as to better reproduce structural properties and molecular interaction energies —
is also an improvement over procedures that utilize energy minimization alone
[218,804,805].
Before describing each potential-energy term in turn, we review the fun-
damental molecular motions, called normal modes, that form the basis for
parameterization of bonded deformations.
9.2 Normal Modes
9.2.1 Quantifying Characteristic Motions
Molecular vibrational spectra of small molecules form the basis for deriving var-
ious force constants, internuclear distances, and bond dissociation energies for
bonded nuclei [766]. Such bond motions describe vibrations about equilibrium
states. Specifically, all possible vibrations of a molecule can be described as a
superposition of the fundamental oscillations (termed normal modes) for that mol-
ecule. Each molecule of N atoms has 3N −6 normal modes: 3 degrees of freedom
per atom (giving 3N ) minus 3 translational and 3 rotational degrees of freedom
for the molecule as a whole.
268 9. Force Fields
Experimental Determination
The vibrational energy levels of molecules can be detected experimentally by
spectroscopic techniques, for example through the vibrational absorption of
infrared radiation (IR) and by Raman scattering.
IR spectroscopy is a powerful technique that captures information on the
transitions between vibrational quantum states, since these transitions lead to
absorption and emission of infrared radiation. The IR wavelength range is
1–100 μm; this spectral range can be compared with the shorter wavelength of the
visible spectrum, which has the range 400–750 nm. IR transitions are ‘allowed’ if
there is a change in the dipole moment of the molecule during the transition.
The complementary technique of Raman spectroscopy captures transitions be-
tween vibrational levels, but the selection rules are different compared to IR:
Raman bands appear only if there is a change in the polarizability of a system
during the transition.
IR absorption and Raman spectroscopy are complementary techniques since
some transitions that have changing dipole moments absorb light, whereas others
have changing polarizability and scatter light. Thus, certain light-induced transi-
tions can be weak, or even absent, in one technique, but of high intensity in the
other. For small symmetric molecules, the observed transitions are complemen-
tary. For larger and asymmetric molecules, however, the selection rules are not
rigidly obeyed, and Raman and IR spectra are essentially the same.
Frequency Units
Rather than expressing these frequencies in inverse seconds (or hertz, Hz, units),
these fundamental frequencies are typically reported in wavenumbers of inverse
centimeters, that is, the number of waves per centimeter. The higher the frequency,
the more difficult (i.e., energetically costly) the deformation. For this reason,
bond-stretching modes generally have higher frequencies than angle-bending
modes, which in turn have higher frequencies than torsion-angle modes.
Note that stretching a bond significantly amounts to breaking it, so the en-
ergy is high; angle bending can be considered to have smaller effects on bonding
(hence energy barriers are lower); torsion barriers are small for rotations about
single bonds since the effects on bonding are smaller still. For double bonds, tor-
sional motion corresponds to bond breaking and the frequencies are higher than
for torsional barriers about single bonds.
Illustration
For larger molecules and complex mixtures, vibrational spectroscopy is a pow-
erful analytical technique for determining which chemical groups are present. To
illustrate, Figure 9.1 shows the three fundamental vibrations of a water mole-
cule: an asymmetric stretch (around 3750 cm
−1
), a symmetric stretch (around
3650 cm
−1
), and an angle-bending mode around 1600 cm
−1
. The symmetric
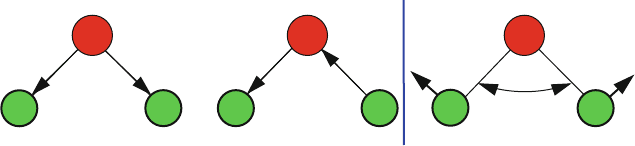
9.2. Normal Modes 269
υ
1
= 3657 cm
−1
(symmetric stretch)
υ
3
= 1595 cm
−1
(bend)
υ
2
= 3776 cm
−1
(asymmetric stretch)
HHHHHH
O
O
O
Figure 9.1. Normal modes of a water molecule.
stretch describes the contraction or elongation of both O–H bonds in concert,
while the asymmetric mode involves this stretching motion in alternate fashion.
The latter has a higher vibrational frequency than the symmetric mode (by about
100 wavenumbers) since the asymmetric vibration is slightly more energetically
costly. The more facile angle-bending deformation has the lowest frequency in
water among these three modes.
9.2.2 Complex Biomolecular Spectra
As the number of atoms in a molecule increases, so does the number of modes,
as well as the associated complexity of the vibrational spectrum. Assigning nor-
mal modes to observed peaks in the experimental spectra becomes more difficult.
Help is available from the characteristic modes of small molecules, which serve
as excellent references for interpretation. In addition, the intensities in the vibra-
tional spectrum can be calculated, at least to a rough approximation,by theoretical
techniques [767].
While vibrational frequencies for the same bond type (e.g., O–H) vary depend-
ing on the molecular context, general values can be assigned to basic two-atom
and three-atom sequences separated into distinct bond types (single, double,
hydrogen-bonded, etc.).
For example, the symmetric O–H stretch in one water molecule (water vapor)
is about 50 wavenumbers higher than that in the H–O–Cl molecule, but 300
wavenumbers higher than the O–H stretching frequency of a hydrogen-bonded
water molecule in liquid water and ice (O–H ···O), where ν = 3400 cm
−1
.This
reduction is due to the attractive force acting on the hydrogen atom in hydrogen-
bonded species, since such attraction reduces the energy and hence the frequency
of the O–H stretching motion.
9.2.3 Spectra As Force Constant Sources
Such spectroscopic measurements and analyses are used to derive appropriate
force constants for biomolecular force fields. Tables 9.1 and 9.2 display exam-
ples of approximate stretching (Table 9.1) and bending and torsional (Table 9.2)
frequencies.