Berg J.M., Tymoczko J.L., Stryer L. Biochemistry
Подождите немного. Документ загружается.

biological systems are catalyzed by dioxygenases, a class of enzymes discovered by Osamu Hayaishi. The active sites of
these enzymes contain iron that is not part of heme or an iron- sulfur cluster.
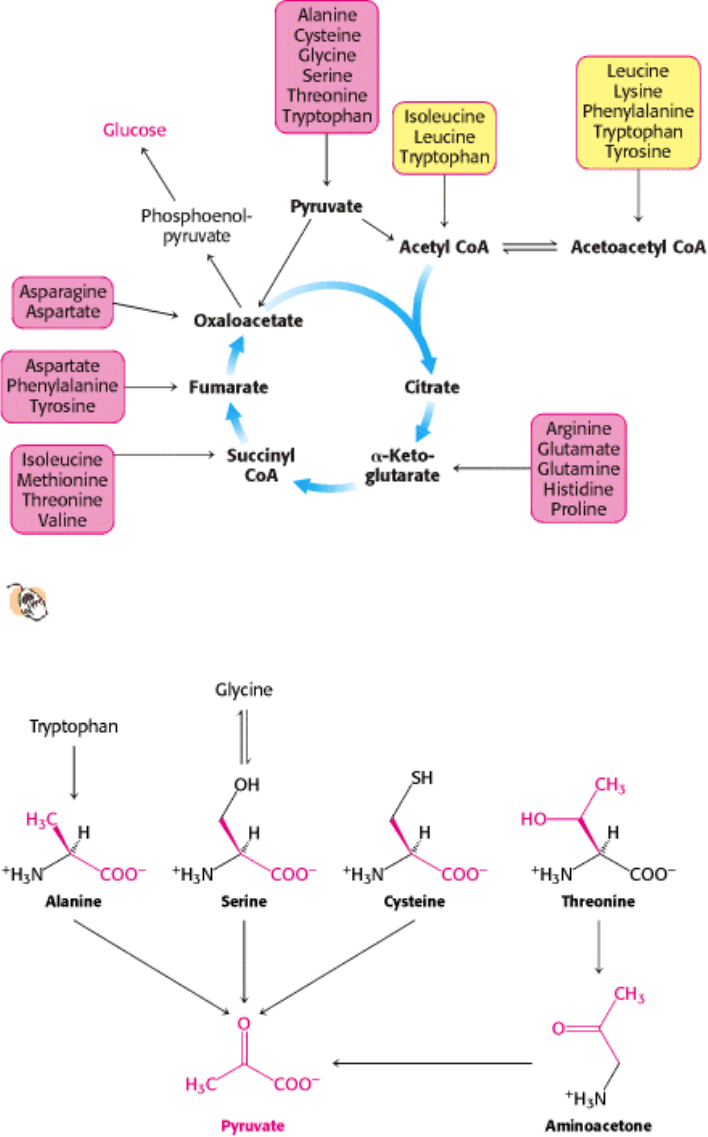
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.21. Fates of the Carbon Skeletons of Amino Acids.
Glucogenic amino acids are shaded red, and ketogenic
amino acids are shaded yellow. Most amino acids are both glucogenic and ketogenic.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.22. Pyruvate Formation from Amino Acids. Pyruvate is the point of entry for alanine, serine, cysteine,
glycine, threonine, and tryptophan.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
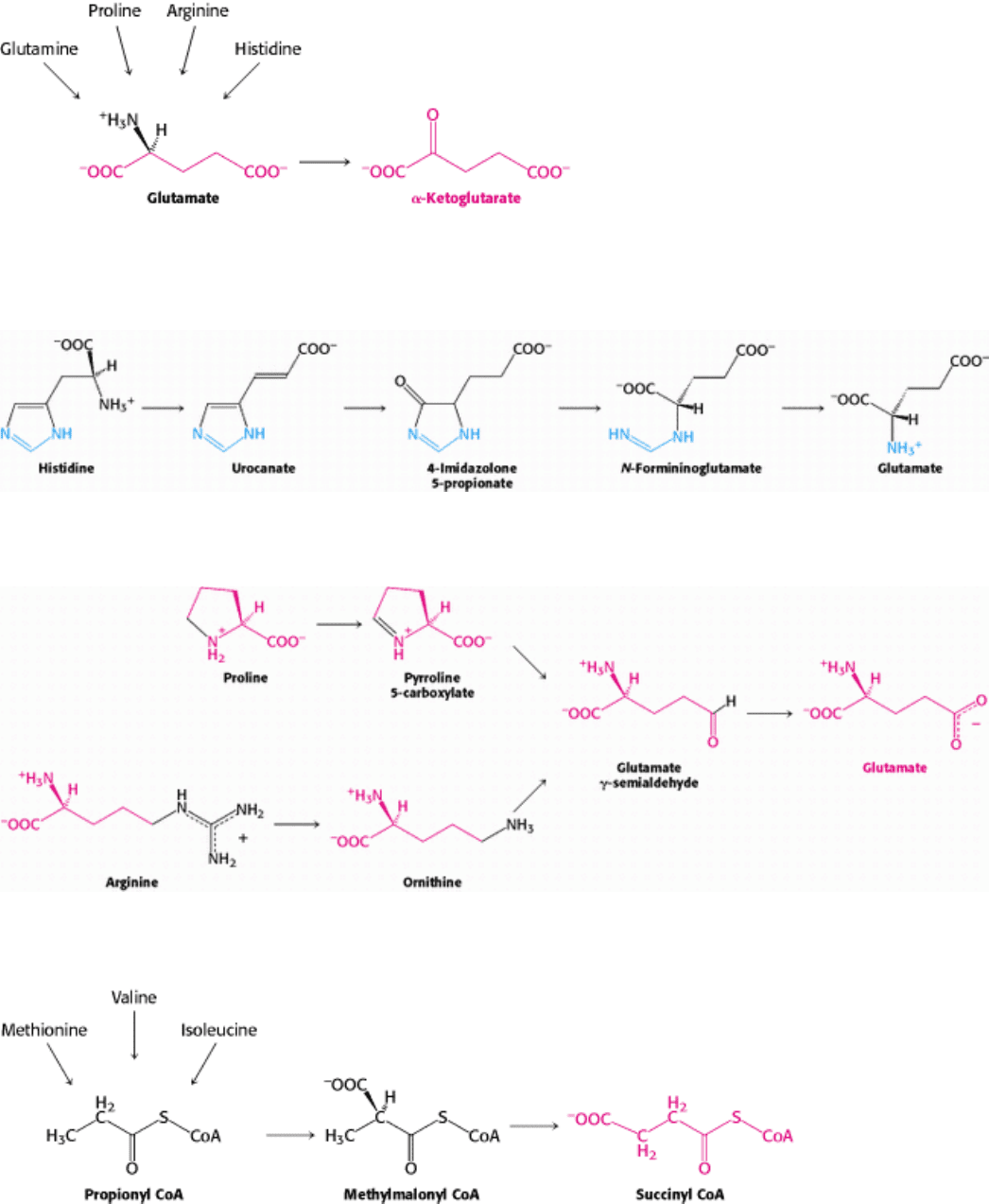
Figure 23.23. α -Ketoglutarate formation from amino acids. α-Ketoglutarate is the point of entry of several five-
carbon amino acids that are first converted into glutamate.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.24. Histidine Degradation. Conversion of histidine into glutamate.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.25. Glutamate Formation. Conversion of proline and arginine into glutamate.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.26. Succinyl CoA Formation. Conversion of methionine, isoleucine, and valine into succinyl CoA.

II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.27. Methionine Metabolism. The pathway for the conversion of methionine into succinyl CoA. S-
Adenosylmethionine, formed along this pathway, is an important molecule for transferring methyl groups.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.28. Formation of Tetrahydrobiopterin, an Important Electron Carrier. Tetrahydrobiopterin can be
formed by the reduction of either of two forms of dihydrobiopterin.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
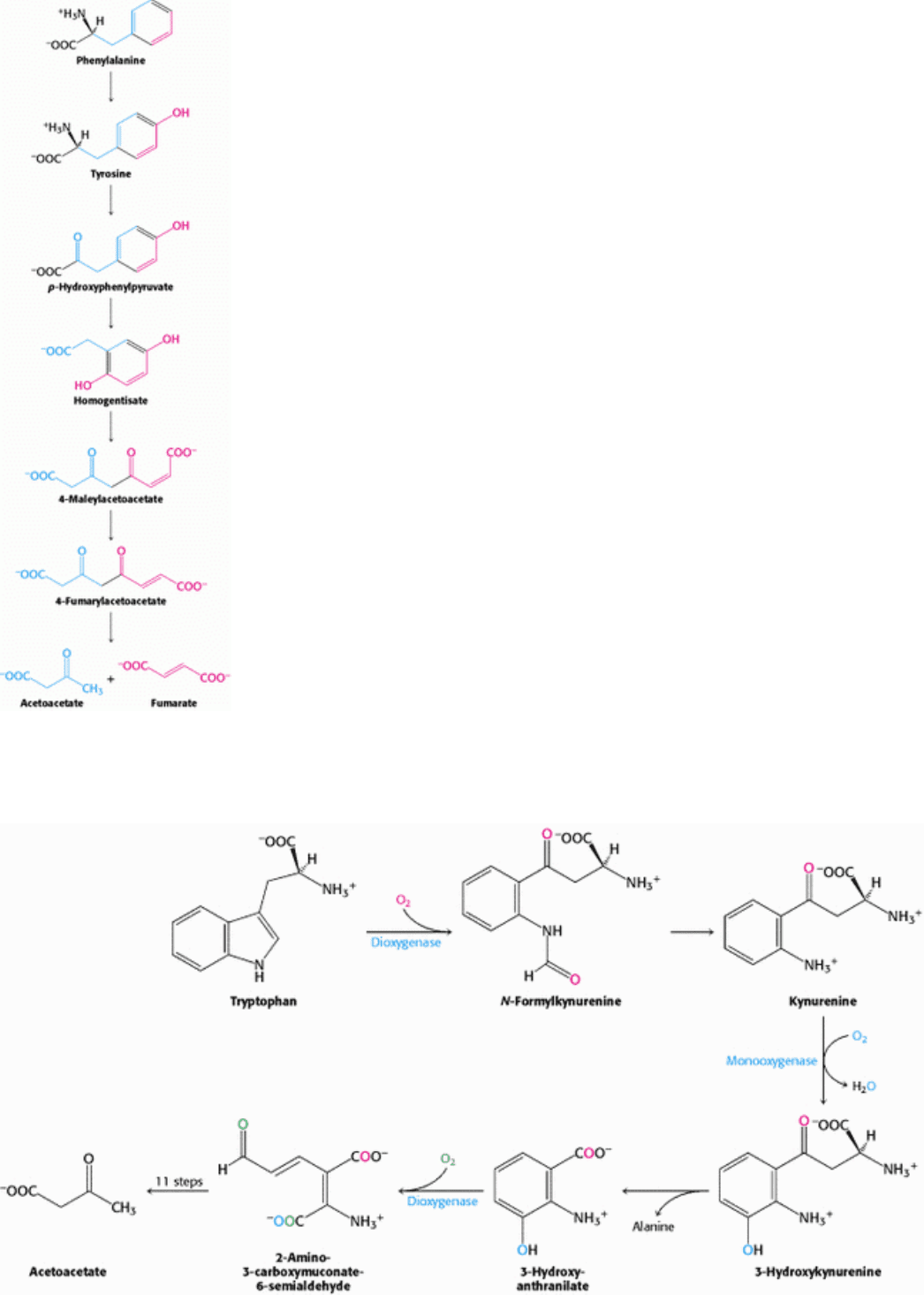
Figure 23.29. Phenylalanine and Tyrosine Degradation. The pathway for the conversion of phenylalanine into
acetoacetate and fumarate.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
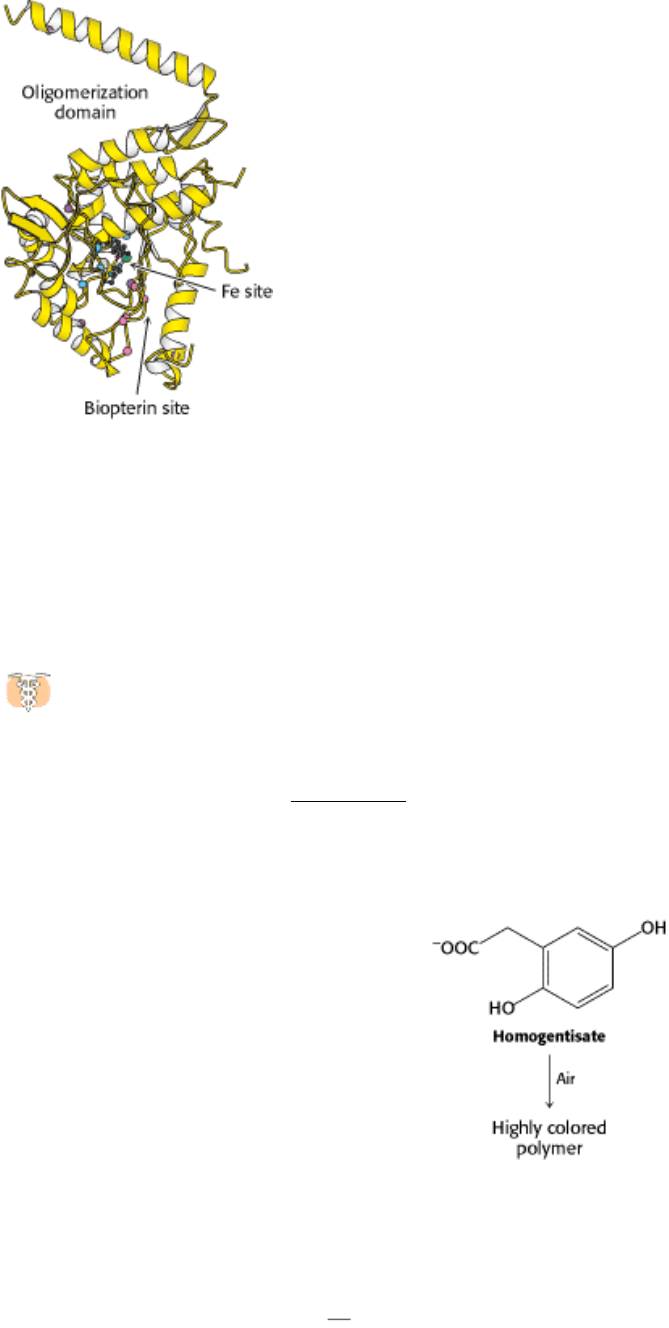
Figure 23.30. Tryptophan Degradation. The pathway for the conversion of tryptophan into alanine and acetoacetate.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.5. Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
Figure 23.31. Structure of One Subunit of Phenylalanine Hydroxylase. Mutations in the genes encoding this enzyme
cause phenylketonuria. More than 200 point mutations have been identified in these genes. The positions of five
mutations affecting the active site (blue), the biopterin-binding site (red), and other regions of the protein (purple) are
indicated as colored spheres.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism
23.6. Inborn Errors of Metabolism Can Disrupt Amino Acid Degradation
Errors in amino acid metabolism provided some of the first correla- tions between biochemical defects and
pathological conditions. For instance, alcaptonuria is an inherited metabolic disorder caused by the absence of
homogentisate oxidase. In 1902, Archibald Garrod showed that alcaptonuria is transmitted as a single recessive
Mendelian trait. Furthermore, he recognized that homogentisate is a normal intermediate in the degradation of
phenylalanine and tyrosine (see Figure 23.29) and that it accumulates in alcaptonuria because its degradation is blocked.
He concluded that "the splitting of the benzene ring in normal metabolism is the work of a special enzyme, that in
congenital alcaptonuria this enzyme is wanting." Homogentisate accumulates and is excreted in the urine, which turns
dark on standing as homogentisate is oxidized and polymerized to a melanin-like substance.
Although alcaptonuria is a relatively harmless condition, such is not the case with other errors in amino acid metabolism.
In maple syrup urine disease, the oxidative decarboxylation of α-ketoacids derived from valine, isoleucine, and leucine
is blocked because the branched-chain dehydrogenase is missing or defective. Hence, the levels of these α-ketoacids and
the branched-chain amino acids that give rise to them are markedly elevated in both blood and urine. Indeed, the urine of
patients has the odor of maple syrup
hence the name of the disease (also called branched-chain ketoaciduria). Maple
syrup urine disease usually leads to mental and physical retardation unless the patient is placed on a diet low in valine,
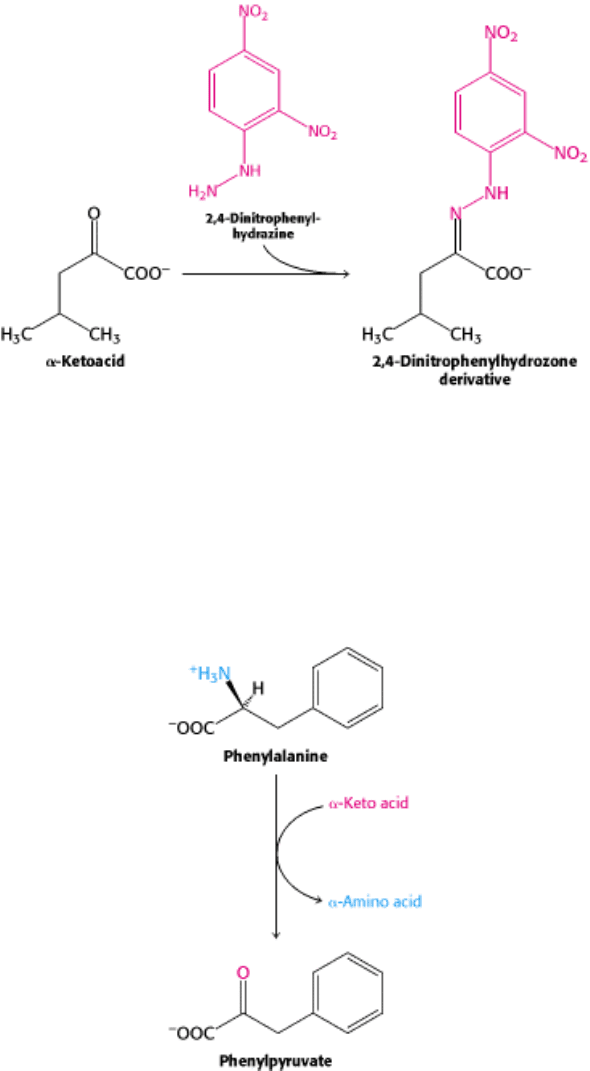
isoleucine, and leucine early in life. The disease can be readily detected in newborns by screening urine samples with 2,4-
dinitrophenylhydrazine, which reacts with α-ketoacids to form 2,4-dinitrophenylhydrazone derivatives. A definitive
diagnosis can be made by mass spectrometry.
Phenylketonuria is perhaps the best known of the diseases of amino acid metabolism. Phenylketonuria is caused by an
absence or deficiency of phenylalanine hydroxylase or, more rarely, of its tetrahydrobiopterin cofactor. Phenylalanine
accumulates in all body fluids because it cannot be converted into tyrosine. Normally, three-quarters of the
phenylalanine is converted into tyrosine, and the other quarter becomes incorporated into proteins. Because the major
outflow pathway is blocked in phenylketonuria, the blood level of phenylalanine is typically at least 20-fold as high as in
normal people. Minor fates of phenylalanine in normal people, such as the formation of phenylpyruvate, become major
fates in phenylketonurics.
Indeed, the initial description of phenylketonuria in 1934 was made by observing the reaction of phenylpyruvate with
FeCl
3
, which turns the urine olive green. Almost all untreated phenylketonurics are severely mentally retarded. In fact,
about 1% of patients in mental institutions have phenylketonuria. The brain weight of these people is below normal,
myelination of their nerves is defective, and their reflexes are hyperactive. The life expectancy of untreated
phenylketonurics is drastically shortened. Half are dead by age 20 and three-quarters by age 30. The biochemical basis of
their mental retardation is an enigma.
Phenylketonurics appear normal at birth, but are severely defective by age 1 if untreated. The therapy for
phenylketonuria is a low phenylalanine diet. The aim is to provide just enough phenylalanine to meet the needs for

growth and replacement. Proteins that have a low content of phenylalanine, such as casein from milk, are hydrolyzed and
phenylalanine is removed by adsorption. A low phenylalanine diet must be started very soon after birth to prevent
irreversible brain damage. In one study, the average IQ of phenylketonurics treated within a few weeks after birth was
93; a control group treated starting at age 1 had an average IQ of 53.
Early diagnosis of phenylketonuria is essential and has been accomplished by mass screening programs. The
phenylalanine level in the blood is the preferred diagnostic criterion because it is more sensitive and reliable than the
FeCl
3
test. Prenatal diagnosis of phenylketonuria with DNA probes has become feasible because the gene has been
cloned and many mutations have been pinpointed to sites in the protein (see Figure 23.31). Interestingly, whereas some
mutations affect the activity of the enzyme, others do not affect the activity itself but, instead, decrease the enzyme
concentration. These mutations lead to degradation of the enzyme, at least in part by the ubiquitin-proteasome pathway.
The incidence of phenylketonuria is about 1 in 20,000 newborns. The disease is inherited in an autosomal recessive
manner. Heterozygotes, who make up about 1.5% of a typical population, appear normal. Carriers of the phenylketonuria
gene have a reduced level of phenylalanine hydroxylase, as indicated by an increased level of phenylalanine in the blood.
However, this criterion is not absolute, because the blood levels of phenylalanine in carriers and normal people overlap
to some extent. The measurement of the kinetics of the disappearance of intravenously administered phenylalanine is a
more definitive test for the carrier state. It should be noted that a high blood level of phenylalanine in a pregnant woman
can result in abnormal development of the fetus. This is a striking example of maternal-fetal relationships at the
molecular level. Table 23.3 lists some other diseases of amino acid metabolism.
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism 23.6. Inborn Errors of Metabolism Can Disrupt Amino Acid Degradation
Table 23.3. Inborn errors of amino acid metabolism
Disease Enzyme deficiency Symptoms
Citrullinema Arginosuccinate lyase Lethergy, siezures, reduced muscle tension
Tyrosinemia Various enzymes of tyrosine degradation Weakness, self-mutilation, liver damage, mental
retardation
Albinism Tyrosinase Absence of pigmentation
Homocystinuria
Cystathionine β-synthase
Scoliosis, muscle weakness, mental retardation, thin
blond hair
Hyperlysinemia
α-Aminoadipic semialdehyde dehydrogenase
Seizures, mental retardation, lack of muscle tone, ataxia
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism
Summary
Proteins Are Degraded to Amino Acids
Dietary protein is digested in the intestine, producing amino acids that are transported throughout the body. Cellular
proteins are degraded at widely variable rates, ranging from minutes to the life of the organism.
Protein Turnover Is Tightly Regulated
The turnover of cellular proteins is a regulated process requiring complex enzyme systems. Proteins to be degraded are
conjugated with ubi-quitin, a small conserved protein, in a reaction driven by ATP hydrolysis. The ubiquitin conjugating
system is composed of three distinct enzymes. A large, barrel-shaped complex called the proteasome digests the
ubiquitinated proteins. The proteasome also requires ATP hydrolysis to function. The resulting amino acids provide a
source of precursors for protein, nucleotide bases, and other nitrogenous compounds.
The First Step in Amino Acid Degradation Is the Removal of Nitrogen
Surplus amino acids are used as metabolic fuel. The first step in their degradation is the removal of their α-amino groups
by transamination to an α-ketoacid. Pyridoxal phosphate is the coenzyme in all aminotransferases and in many other
enzymes catalyzing amino acids transformations. The α-amino group funnels into α-ketoglutarate to form glutamate,
which is then oxidatively deaminated by glutamate dehydrogenase to give NH
4
+
and α-ketoglutarate. NAD
+
or NADP
+
is the electron acceptor in this reaction.
Ammonium Ion Is Converted into Urea in Most Terrestrial Vertebrates
The first step in the synthesis of urea is the formation of carbamoyl phosphate, which is synthesized from CO
2
, NH
4
+
,
and two molecules of ATP by carbamoyl phosphate synthetase. Ornithine is then carbamoylated to citrulline by orthinine
transcarbamoylase. These two reactions take place in mitochondria. Citrulline leaves the mitochondrion and condenses
with aspartate to form argininosuccinate, which is cleaved into arginine and fumarate. The other nitrogen atom of urea
comes from aspartate. Urea is formed by the hydrolysis of arginine, which also regenerates ornithine. Some enzymatic
deficiencies of the urea cycle can be bypassed by supplementing the diet with arginine or compounds that form
conjugates with glycine and glutamine.
Carbon Atoms of Degraded Amino Acids Emerge as Major Metabolic Intermediates
The carbon atoms of degraded amino acids are converted into pyruvate, acetyl CoA, acetoacetate, or an intermediate of
the citric acid cycle. Most amino acids are solely glucogenic, two are solely ketogenic, and a few are both ketogenic and
glucogenic. Alanine, serine, cysteine, glycine, threonine, and tryptophan are degraded to pyruvate. Asparagine and
aspartate are converted into oxaloacetate. α-Ketoglutarate is the point of entry for glutamate and four amino acids
(glutamine, histidine, proline, and arginine) that can be converted into glutamate. Succinyl CoA is the point of entry for
some of the carbon atoms of three amino acids (methionine, isoleucine, and valine) that are degraded through the
intermediate methylmalonyl CoA. Leucine is degraded to acetoacetyl CoA and acetyl CoA. The breakdown of valine
and isoleucine is like that of leucine. Their α-ketoacid derivatives are oxidatively decarboxylated by the branched-chain
α-ketoacid dehydrogenase.
The rings of aromatic amino acids are degraded by oxygenases. Phenylalanine hydroxylase, a monooxygenase, uses
tetrahydrobiopterin as the reductant. One of the oxygen atoms of O
2
emerges in tyrosine and the other in water.
Subsequent steps in the degradation of these aromatic amino acids are catalyzed by dioxygenases, which catalyze the
insertion of both atoms of O
2
into organic products. Four of the carbon atoms of phenylalanine and tyrosine are
converted into fumarate, and four emerge in acetoacetate.
Inborn Errors of Metabolism Can Disrupt Amino Acid Degradation
Errors in amino acid metabolism served as sources of some of the first insights into the correlation between pathology
and biochemistry. Although there are many hereditary errors of amino acid metabolism, phenylketonuria is the best
known. This condition is the result of the accumulation of high levels of phenylalanine in the body fluids. By unknown
mechanisms, this accumulation results in mental retardation unless the afflicted are placed on low phenylalanine diets
immediately after birth.
Key Terms
ubiquitin

proteasome
aminotransferase (transaminase)
glutamate dehydrogenase
pyridoxal phosphate (PLP)
pyridoxamine phosphate (PMP)
alanine cycle
urea cycle
carbamoyl phosphate synthetase
N-acetylglutamate
ketogenic amino acid
glucogenic amino acid
biopterin
phenylketonuria
II. Transducing and Storing Energy 23. Protein Turnover and Amino Acid Catabolism
Problems
1.
Wasted energy? Protein hydrolysis is an exergonic process, yet the 26S proteasome is dependent on ATP hydrolysis
for activity.
(a) Although the exact function of the ATPase activity is not known, suggest some likely functions.
(b) Small-peptides can be hydrolyzed without the expenditure of ATP. How does this information concur with your
answer to part a?
See answer

2.
Keto counterparts. Name the α-ketoacid that is formed by transamination of each of the following amino acids:
(a) Alanine
(b) Aspartate
(c) Glutamate
(d) Leucine
(e) Phenylalanine
(f) Tyrosine
See answer
3.
A versatile building block. (a) Write a balanced equation for the conversion of aspartate into glucose through the
intermediate oxaloacetate. Which coenzymes participate in this transformation? (b) Write a balanced equation for the
conversion of aspartate into oxaloacetate through the intermediate fumarate.
See answer
4.
The benefits of specialization. The archaeal proteasome contains 14 identical active β subunits, whereas the
eukaryotic proteasome has 7 distinct β subunits. What are the potential benefits of having several distinct active
subunits?
See answer
5.
Propose a structure. The 19S subunit of the proteasome contains 6 subunits that are members of the AAA ATPase
family. Other members of this large family are associated into homohexamers with six-fold symmetry. Propose a
structure for the AAA ATPases within the 19S proteasome. How might you test and refine your prediction?
See answer
6.
Effective electron sinks. Pyridoxal phosphate stabilizes carbanionic intermediates by serving as an electron sink.
Which other prosthetic group catalyzes reactions in this way?
See answer
7.
Helping hand. Propose a role for the positively charged guanidinium nitrogen atom in the cleavage of
argininosuccinate into arginine and fumarate.
See answer
8.
Completing the cycle. Four high-transfer-potential phosphoryl groups are consumed in synthesizing urea according
to the stoichiometry given in Section 23.4.2. In this reaction, aspartate is converted into fumarate. Suppose that
fumarate is converted back into aspartate. What is the resulting stoichiometry of urea synthesis? How many high-
transfer-potential phosphoryl groups are spent?
See answer