Caballero B. (ed.) Encyclopaedia of Food Science, Food Technology and Nutrition. Ten-Volume Set
Подождите немного. Документ загружается.


been identified, which underscores the complexity of
nutrient–gene regulation.
0021 In contrast, an indirect effect may be exerted by
some nutrient metabolites through secondary metab-
olism. The effect of dietary fiber is a well-studied
example of indirect gene modulation. Intestinal bac-
teria metabolize undigested fiber and release butyric
acid. Although the protective action of butyric acid
on colonocytes has not been fully elucidated, in vitro
studies suggest that butyric acid may modulate gene
expression by direct and indirect means. It may acet-
ylate or phosphorylate histones in the nucleus, which
would affect the binding of transcription factors to
regulatory elements of genes. In particular, elevated
butyrate has been shown to inhibit histone deacety-
lase, which results in the upregulation and secretion
of IL-8, a potent chemotactic factor for neutrophils.
In addition, butyrate could also act as an oxidative
substrate, which would modulate cellular signaling
networks via G-proteins. Investigation of these mech-
anisms may uncover new dietary treatments and
better understanding of the progression of colorectal
cancer. Moreover, butyrate-producing bacteria in the
intestine can vary depending on the diet. Infants fed a
casein-based formula predominantly excrete butyric
acid and propionic acid, in contrast to breast-fed
infants whose stool contains mostly acetic acid.
Thus, the presence of butyrate can relay signals to
the mucosal immune system, providing vital informa-
tion about luminal contents and events.
0022 Expression of genes for proteins involved in anti-
gen presentation, namely major histocompatibility
complex (MHC) class II molecules and the invariant
chain Ii on enterocytes, are also regulated by diet. In
mice, prolonged exposure to dams’ milk delayed the
expression of MHC class II and Ii molecules, suggest-
ing that early oral dosing of antigens could modify the
level of antigen presentation. Furthermore, the ap-
pearance of MHC class II and Ii molecules was
delayed in mice fed an elemental diet composed of
synthetic amino acids and fats as compared to litter-
mates fed standard cereal-based rodent diets. Thus,
gene expression in intestinal epithelial cells can be
manipulated via the presence of specific dietary
proteins in the gut lumen.
0023 The complexity of nutrient–gene interactions in-
volves not only interaction with the genome and
gene transcription but also affects posttranscriptional
events. The regulatory factors involved in posttran-
scriptional control may be the dietary components
themselves, metabolites, or changes in hormones,
making it difficult to distinguish the regulatory path-
way. There are some examples of nutritional modifi-
cations where the regulatory sequences that are
targeted are from 3
0
and 5
0
untranslated regions
(UTR) of mRNA that bind specific proteins, a process
that in turn regulates polyadenylation, translation,
localization, and stability of mRNA. It may be pos-
sible in the future to define nutrient–gene interactions
in order to tailor dietary requirements to individuals
with different disease risk profiles.
Immunonutrition
0024Lessons learned from studying the immunomodula-
tory potential of individual nutrients in patients
undergoing surgery or recovering from various tissue
damage have led to the development of a therapeutic
approach called immunonutrition. These special ent-
eral diets are formulated with components that have
been shown to enhance or preserve host immune
reactions, or suppress exaggerated inflammatory re-
sponses, with the aim of decreasing the morbidity and
mortality of critically ill patients. Immune-enhancing
enteral diets have been assessed in 12 prospective,
randomized clinical trials. Although these studies
show a strong and statistically significant benefit to
most patients, the ideal composition of such a diet
still needs to be established. Furthermore, a better
definition of the type of patient who would benefit
most from immunonutrition is needed before this
therapy is widely adopted.
Autoimmunity
0025Nutrition-based therapies of autoimmune disease are
a novel approach that exploits the immunomodula-
tory nature of certain foods. For example, vitamins
have been reported to act as immune regulators in
multiple sclerosis. 1,25-Dihydroxy vitamin D
3
sup-
plementation of mice with experimental autoimmune
encephalomyelitis (EAE), a murine model of multiple
sclerosis, prevented the progression of the disease,
while withdrawal caused regression to multiple scler-
osis-like symptoms. Nutrition-based therapy using
gamma-linolenic acid (18:3n-6) supplementation has
also reduced the signs and symptoms of disease activ-
ity in rheumatoid arthritis. Similarly, dietary modifi-
cation has been shown significantly to inhibit the
development of diabetes in both rodent models of
spontaneous autoimmune diabetes, BioBreeding rats
and nonobese diabetic (NOD) mice. It was shown that
hydrolyzed casein diets had less diabetes-inducing
potential than cereal-based diets. Of particular inter-
est, it was also observed that early oral exposure to
antigens from known diabetes-promoting diets
delayed and inhibited development of diabetes in
about one-third of neonates. The identity of the
immunomodulatory agent(s) in the diet is currently
being investigated.
3260 IMMUNOLOGY OF FOOD

0026 In other inflammatory disorders of the human
intestine, such as celiac disease, ulcerative colitis,
and Crohn’s disease, a cytokine imbalance may
trigger and potentiate the destructive process. In the
case of celiac disease, the gut immune system is
sensitized to certain wheat peptides. Upon ingestion
of wheat, individuals with certain disease-risk haplo-
types, particularly the antigen-presenting molecules
human leukocyte antigen (HLA)-DQ2 and HLA-
DQ8, may display gliadin proteins in a conformation
that is recognized by lamina propria T cells. Once
activated, these T cells could initiate a destructive
inflammatory process that releases the endomysial
autoantigen, tissue transglutaminase, and thus po-
tentiates the immune attack and atrophy of intestinal
villi. When wheat proteins are eliminated from the
diet, the immune attack stops and the symptoms dis-
appear. Of note, is the observation that celiac disease
predisposes individuals to other autoimmune dis-
orders such as type 1 diabetes, dermatitis herpeti-
formis, autoimmune thyroiditis, collagen diseases,
autoimmune alopecia, and autoimmune hepatitis.
This correlation is stronger in cases of long-term
undiagnosed and untreated celiac disease, further
suggesting that the GALT is an important site for
immune regulation and potential stimulation of auto-
reactive cells.
New Technologies
0027 New approaches are being explored to assess the
immunomodulatory nature of food components. For
instance, genomic analysis using microarray chip
technology can detect differences in the level of
transcription of genes. Alternatively, protein mixtures
can be characterized using two-dimensional electro-
phoresis and mass spectrometry to obtain amino acid
sequences of proteins that can be matched with those
in existing databases. This approach can be combined
with immunoblotting using sera from susceptible
individuals or specific monoclonal antibodies. Such
proteomic analysis has been used to characterize
wheat allergens in baker’s asthma. Since there is
debate over the risks of toxins, allergens, and other
chemical hazards stemming from new food produc-
tion and processing technologies, these modern tools
may be invaluable in food safety assessment. These
may also serve as predictive tools for predicting risk
of food immunogenicity and allergenicity.
0028 Transgenic plants are being examined as vehicles
for vaccine delivery. A case in point is transgenic
lettuce plants expressing hepatitis B virus surface
antigens that were ingested by human volunteers
and resulted in a specific serum-IgG response to
the plant-produced viral proteins. Other examples
include genetically modified potatoes expressing
Norwalk virus capsid protein or autoantigen for
autoimmune therapy. Transgenic plants may serve as
an inexpensive alternative to fermentation systems
for production of antigens. They may also avoid
costly purification processes because the subject
ingests directly the modified plant. Oral vaccination
with bioengineered plants is currently being evalu-
ated for long-term safety, effectiveness, and cost-
effectiveness. Transgenic tobacco plants containing
the autoantigen GAD-65 (glutamic acid decarboxy-
lase-65) were fed to diabetes-prone NOD mice and
this treatment prevented animals from developing
diabetes. These data open the possibility of using
bioengineered plants as vehicles for the delivery of
molecules that may be tolerogenic.
Summary
0029The nutrition recommendations that are currently
suggested by government agencies and health profes-
sionals are based on epidemiological studies of dis-
crete populations. The weakness of these studies is
that they do not take into account our inherent bio-
chemical and genetic individuality, as was already
recognized in 1956 by Williams. The food we eat
brings us into direct contact with a complex mixture
of nutrients, nonnutrients, toxins, microbes, and
molecules that are similar to those that make up our
own bodies. Many of these have biological activity
that can modify host immune system reactivity. This
multitude of food components can alter the nutri-
tional milieu, provide antigens that must be dealt
with, act as modifiers of lymphoid cell membranes,
alter the balance of immune regulatory (or other)
cell compounds such as growth factors, hormones,
metabolites, cytokines, chemokines, inflammatory
mediators, or interact directly with the genes that
are important controllers of the immune response.
We are at a stage when new tools for high-throughput
screening and analysis of gene and protein expression
are providing opportunities for understanding com-
plex interactions among food, genes, and proteins.
These technologies and our increasing ability to
categorize and comprehend the large amounts of
information they generate will permit new insights
into the mechanisms by which our exposure to hun-
dreds of chemicals from foods affects our immune
systems. This will eventually permit us to under-
stand the range of immune system reactivity and
how it is influenced by foods we eat as well as other
environmental factors. The advent of genome
and proteome exploration may yield information
that will permit nutrition-based therapies designed
for individuals.
IMMUNOLOGY OF FOOD 3261

Acknowledgements
0030 The authors thank the following agencies for sup-
porting the research program in Dr. Scott’s labora-
tory: Juvenile Diabetes Research Foundation (JDF),
Canadian Inistitutes of Health Research (CIHR),
Ontario Research and Development Challenge
Fund, Canada Founation for Innovation, Health
Canada. Karolina Burghardt was the recipient of a
scholarship from the Diabetic Childrens Foundation
and the CIHR.
See also: Anemia (Anaemia): Iron-deficiency Anemia;
Copper: Properties and Determination; Physiology;
Essential Fatty Acids; Fats: Classification; Food
Intolerance: Types; Food Allergies; Milk Allergy; Lactose
Intolerance; Elimination Diets; Garlic; Infants: Breast-
and Bottle-feeding; Nutrition Education; Selenium:
Properties and Determination; Zinc: Deficiency
Further Reading
Brandtzaeg P (1998) Development and basic mechanisms of
human gut immunity. Nutrition Reviews 56: S5–S18.
Cousins RJ (1999) Nutritional regulation of gene expres-
sion. American Journal of Medicine 106(1A): 20S–23S.
Cunningham-Rundles S (2001) Nutrition and the mucosal
immune system. Current Opinion in Gastroenterology
17: 171–176.
Hanson LA (1999) Human milk and host defense: immedi-
ate and long-term effects. Acta Paediatrica 88(430)
(Suppl.): 42–46.
Hesketh JE, Vasconcelos MH and Bermano G (1998) Regu-
latory signals in messenger RNA: determinants of
nutrient–gene interaction and metabolic compartmenta-
tion. British Journal of Nutrition 80: 307–321.
Heyman M (1999) Evaluation of the impact of food tech-
nology on the allergenicity of cow’s milk proteins. Pro-
ceedings of Nutritional Society 58: 587–592.
Koletzko B, Aggett PJ, Bindels JG et al. (1998) Growth,
development and differentiation: a functional food sci-
ence approach. British Journal of Nutrition 80(suppl. 1):
S5–S45.
Lamm DL and Riggs DR (2001) Enhanced immunocompe-
tence by garlic: role in bladder cancer and other malig-
nancies. Journal of Nutrition 131: 1067S–1070S.
Scott FW (1996) Food induced Type 1 diabetes in the BB
rat. Diabetes Metabolism Reviews 12: 341–359.
Smith KM, Eaton AD, Finlayson LM and Garside P (2000)
Oral tolerance. American Journal of Respiratory Critical
Care Medicine 162: S175–S178.
Strobel S and Mowat AM (1998) Immune responses
to dietary antigens: oral tolerance. Immunology Today
19: 173–181.
Voelker R (2000) The hygiene hypothesis. Journal of the
American Medical Association 283: 1282.
Weindruch R, Kayo T, Lee CK and Prolla TA (2001) Micro-
array profiling of gene expression in aging and its alter-
ation by caloric restriction in mice. Journal of Nutrition
131: 918S–923S.
Williams RJ (1956) Biochemical variations; its significance
in biology and medicine. In: Biochemical Individuality,
pp. 1–7. New York: Wiley.
Ziboh VA (2000) Nutritional modulation of inflammation
by polyunsaturated fatty acids/eicosanoids. In: Gersh-
win ME, German JB and Keen CL (eds) Nutrition and
Immunology: Principles and Practice, pp. 157–167.
Totowa, NJ: Humana Press.
INBORN ERRORS OF METABOLISM
Overview
K de Meer, Vrije Universiteit Medical Center,
Amsterdam, The Netherlands
Copyright 2003, Elsevier Science Ltd. All Rights Reserved.
Scope
0001 In clinical medicine and food science, metabolic dis-
orders form a small but important field. Removal of
nutrients which can have a critical effect on develop-
ment of toxic crises or irreversible detrimental effect
on organ function is a potentially simple and some-
times life-saving treatment. Simple measures, such as
avoidance of prolonged fasting, can be very effective
in selected cases. Special diets and costly supplements
are necessary in others. Although inborn errors of
metabolism are collectively numerous, most disorders
are individually rare. Clinical presentation can vary
substantially over the spectrum of inborn errors of
metabolism and patients can be seen at any age by
pediatricians, neurologists, cardiologists, immunolo-
gists, hepatologists, dermatologists, and gynecolo-
gists. There is no common single test to screen for
the whole group of diseases. Collaboration between
specialized clinicians and biochemists, metabolic
laboratories, and the food industry is required for
adequate procedures in the diagnostic work-up and
for proper treatment.
0002Much expert and scientific information on individ-
ual genetic variation in relation to human disease
is available online (Table 1). This article offers a
3262 INBORN ERRORS OF METABOLISM/Overview

conceptual approach to the field, and describes a
selection of diseases in which nutritional intervention
is possible.
Genetics and Inborn Errors of Metabolism
0003 Inherited metabolic diseases cause structural changes
or metabolic derangements in the body. The term
‘inborn error of metabolism’ was presented by John
Garrow in 1906 in a lecture about alcaptonuria, in
which he also hypothesized the theory of one gene,
one protein as its cause. Although the concept of
inborn error of metabolism implies that a deficiency
of a single enzyme is present, the term has also come
into use to describe inherited disorders in proteins not
involved in metabolism. Mutated genes and their
translation products, proteins, can cause derange-
ments during body and tissue formation, and can
also be present as abnormal structural proteins and
diminished enzyme activity in a wide range of pro-
cesses. Autosomal recessive inheritance is the most
common form of inheritance encountered in inborn
errors of metabolism, but not all affected individuals
necessarily have symptoms and other inheritance
patterns are all but rare.
000 4 Mutations in the estimated 30000 human genes
can in principle cause a very large number of diseases.
More than 2200 disease loci are now known. In the
human genome, mutations in a number of genes are
not compatible with survival at the time the embryo
develops. In some cases the same mutation in an allele
is known to be causally related to more than one
disease. Within one disease the same genotype can
be associated with more than one phenotypical pre-
sentation. Given the magnitude of the human
genome, and the multitude of possible mutations
and phenotypic disease states, it is not possible to
recognize all inborn errors of metabolism by neonatal
screening. Apart from dedicated neonatal screening
for the more frequent and treatable disorders, simple
methods using clinical diagnosis and targeted labora-
tory investigations are needed to identify the inborn
error of metabolism in an individual patient.
0005From a pathophysiological point of view, the
metabolic disorders can be divided into a number of
groups that can be used to facilitate diagnostic
decision making, including the initial differential
diagnosis by the general practitioner and specialist.
Specialists in pediatrics, internal medicine, neurology,
clinical chemistry, and genetics collaborate in the
clinical field of metabolic diseases for establishing
the diagnosis on the level of the protein or gene, and
to provide treatment and genetic counseling. The
focus of this chapter is on inborn errors of metabol-
ism in which nutritional modulation is an important,
and in some cases the only, is of treatment. They are
summarized in Table 2.
0006Not all inborn errors of metabolism with nutri-
tional consequences are covered in this article.
Familial hyperlipidemias are, also in terms of numbers
of patients, a very important group of inborn errors
of metabolism, and are dealt with elsewhere in this
encyclopedia. Some inborn errors of metabolism in
which food components play a role, notably glucose-
6-phosphate dehydrogenase deficiency, in which fava
beans can trigger symptoms, and porphyrias in which
low carbohydrate intake can aggravate symptoms,
are not further explored here.
Pathophysiology
0007Metabolic disorders can be divided into groups,
according to the pathophysiological mechanism that
is involved.
tbl0001 Table 1 Inborn errors of metabolism: online information sources
OMIM (Johns Hopkins University, Baltimore)
Extensive information on clinical and molecular topics of inborn errors of metabolism and other inherited diseases is available
fromwww.ncbi.nlm.nih.govintheOnlineMendelianInheritanceinMan(OMIM)library.Itcombinesthehumangenemapwitha
morbidity map. The OMIM database assigns a code (MIM) number, gives the locus symbol, and provides a complete list of
clinical phenotypes for each disease entity. Experts also describe the clinical picture. OMIM provides a morbidity search machine
for differential diagnosis on the basis of clinical symptoms and signs. At present, the number of gene loci with an associated
clinical disease entity exceed 1700, and more than 2250 human phenotypes have been mapped on the genome. In more than 70%
of these disease phenotypes, the molecular basis has been defined. The National Center of Biomedical Information (NCBI) website of
the National Library of Medicine at the National Institute of Health also hosts the medical scientific literature (PubMed) and human
genome (GenBank) library databases. Together these resources provide an intertwined and up-to-date online source for
practitioners and scientists
HUGO Mutation Database Initiative (University of Melbourne)
The website ariel.ucs.unimelb.edu.au/*cotton/mdi.htm provides a list of locus-specific databases according to gene designation,
available from university and research institute sources around the world. Connected by the MIM number of each locus, the
information from OMIM is only one click away. The drawback is that the visitor should already know the locus lettercode prior to
starting up the link. HUGO also lists patient information sites. Information on nonhuman genetic variation is also available
INBORN ERRORS OF METABOLISM/Overview 3263

0008 Group 1 is comprised of disorders characterized by
the disturbed synthesis or catabolism of complex
molecules. Symptoms are not related to food intake,
and are permanent, progressive, and independent of
intercurrent infections. Lysosomal storage disease,
peroxisomal disorders, a
1
-antitrypsin deficiency, and
congenital disorders of glycosylation are in this
group. With some notable exceptions, nutritional
therapy is not effective in these disorders.
0009 Group 2 includes all disorders of intermediate
metabolism. Because multiple enzymes are involved
in the metabolism between macronutrients and end
products, these disorders lead to accumulation of
toxic compounds proximal to the metabolic block.
There can be progressive or acute intoxication due to
these compounds, and intercurrent infections or ex-
cessive intake of foods can lead to a metabolic crisis.
A symptom-free interval is characteristic. Late onset
or an intermittent clinical presentation is typical.
Aminoacidemias, most organic acidurias, urea cycle
defects, and sugar intolerances belong to this group.
0010Group 3 consists of inborn errors involved in inter-
mediate metabolism which is directly related with
cellular energy transfer. Enzyme defects can affect
the supply with macromolecules involved in energy
production as well as the transfer of energy by mol-
ecules in the matrix and in the inner membrane
respiratory chain system within the mitochondria.
Glycogen storage diseases, gluconeogenesis defects,
defects in pyruvate carboxylase and dehydrogenase,
tbl0002 Table 2 Selected metabolic disorders for which nutritional therapy is used
Amino acid disorders
Phenylketonuria Protein restriction, tyrosine and docosa-hexanoic acid supplementation
Tyrosinemia type I and II Protein restriction, NTBC in type I
Maple syrup urine disease Valine, leucine, isoleucine restriction, and thiamin supplementation
Urea cycle defects Protein restriction, arginine and sodium benzoate and phenylacetate
supplementation
Homocysteinuria (CBS-deficiency) Methionine restriction, vitamin B
6
, and betaine supplementation
Isovaleric acidemia Leucine restriction, glycine and carnitine supplementation
Serine synthesis defect (3-PGDHD) Serine and glycine supplementation
Hartnup disease High-protein, nicotinamide supplementation
Carbohydrate disorders
GSD type I and III High carbohydrate/low fat and cholesterol
Galactosemia Galactose (and lactose) restriction
Hereditary fructose intolerance Fructose restriction
Glucose-galactose malabsorption High fructose/low glucose-galactose
Congenital disorder of glycosylation Ib Mannose supplementation
Organic acidemias and acidurias
Propionic acidemia Protein restriction, avoidance of dehydration, Inhibition of colonic flora with
metronidazole
Methyl malonic acidemia Protein restriction, vitamin B
12
supplementation
Multiple acyl coenzyme A dehydrogenase deficiency Fat restriction, riboflavin and carnitine supplementation
Glutaric aciduria type I Lysine restriction, riboflavin supplementation
Fatty acid oxidation and ketolysis defects
b-Ketothiolase deficiency Low protein, avoid fasting
MCAD, LCAD, LCHAD deficiency Low fat, avoid fasting, + carnitine
Mitochondrial disorders
Respiratory-chain disorders Increased energy need in early life
Pyruvate dehydrogenase deficiency High fat/low carbohydrate and thiamin
Micronutrient disorders
Biotinidase deficiency Biotin supplementation
Vitamin B
6
-dependent epilepsy Vitamin B
6
supplementation
Transcobalamin II deficiency Vitamin B
12
supplementation
Immerslund-Grsbeck disease Vitamin B
12
supplementation
Hereditary folate malabsorption Folate supplementation
Methylene tetrahydrofolate reductase deficiency Folate supplementation
Acrodermatitis enterohepatica Zinc supplementation
Wilson disease Zinc supplementation
NTBC, nitro-trifluoro-methylbenzoyl-cyclohexanedione; CBS, cystathionine b-synthase: homozygous patients cannot metabolize homocysteine which is
formed after transmethylation of methionine; 3-PGDHD, 3-phosphoglycerate dehydrogenase deficiency: the enzyme block prevents the biosynthesis of
serine; GSD, glycogen storage disease; MCAD, medium-chain acyl coenzyme A dehydrogenase; LCAD, long-chain acyl coenzyme A dehydrogenase;
LCHAD, long-chain hydroxy acyl coenzyme A dehydrogenase: these mitochondrial enzymes form part of the oxidation of fatty acids.
3264 INBORN ERRORS OF METABOLISM/Overview

fatty acid oxidation defects, and mitochondrial
respiratory chain disorders are present in this group.
0011 Group 4 is comprised of diseases in which disturb-
ances are present in: (1) membrane transport; (2)
intracellular signaling; or (3) critical developmental
periods. Defects of transmembrane transporters for
carbohydrates, amino acids, and lipids in the intestine
are suspected by clinical observation (diarrhea, fail-
ure to thrive) and absorption studies. Similarly,
defects can be present in tubular reabsorption in the
kidney. Other clinical entities are related to disturbed
transport of metals (e.g., of copper, such as Menkes
and Wilson disease). New insights have been de-
veloped that complex compounds, produced in
specialized metabolic tissues and necessary for
specialized functions in the brain, must undergo a
critical step in transport over the cell membrane for
proper function. Similarly, with respect to intracellu-
lar signaling functions, the assembly or metabolism of
complex compounds in the central nervous system
and other organs can be deranged. Defects of intra-
cellular membrane transport comprise another group
of newly recognized disorders. The transport defects
generally result in the dysfunction of one or more
affected organs (e.g., diarrhea in carbohydrate mal-
absorption, liver failure and brain dysfunction by
copper storage in Wilson disease, brain dysfunction
in creatine transporter deficiency, X-linked non-
specific mental retardation, and abnormal neuro-
transmitter synthesis) or deficiency syndromes (due
to malabsorption).
0012 Recently, insights in developmental biology led to
the discovery that certain compounds (e.g., choles-
terol) with known functions during extrauterine life
are critical for developmental gene description in the
embryo (e.g., absence of 7-dehydrocholsterol biosyn-
thesis causes Smith–Lemli–Opitz syndrome, charac-
terized by dysmorphic features and severe mental
retardation). Developments in molecular biology
and the genome project, human genetics, and clinical
chemistry enable discovery of the etiology and patho-
physiology of newly recognized disorders with a
range of clinical presentations.
Clinical and Laboratory Expertise in
Diagnosis
0013 Children with inborn errors of metabolism may pre-
sent with one or more of a large variety of symptoms
and signs. Although most Mendelian phenotypes are
expressed early in life, adult presentations are recog-
nized in increasing numbers. The patient history,
clinical assessment (for color, odor, hepatomegaly,
neurological abnormalities, including hypotonia, and
dysmorphic features), and immediate laboratory
investigations are necessary for initial judgment in
terms of the pathophysiology group and index of
suspicion for an inborn error of metabolism. A full
medical and genetic family history is important and
should include the circumstances of any stillbirth,
sudden infant death, unusual death in childhood or
early adulthood, and information on consanguinity
between the parents.
0014Blood investigations should include routine hema-
tology and electrolytes (search for anion gap), glu-
cose, creatinine, liver enzymes, bilirubin, ammonia,
calcium, phosphate, lactate, pyruvate, ketone bodies
(3-hydroxy- and ketobutyrate), fatty acids, uric
acid, blood-gas analysis, and prothrombin time. Urin-
alysis should include acetone, reducing substances,
pH, sulfite, electrolytes, and uric acid. Further investi-
gation may comprise lumbar puncture, chest X-ray,
and cardiac and central nervous system function
studies. Further laboratory and other investigations
are guided by the pathophysiological group, abnor-
malities in the routine investigations, and suspicion of
an individual disorder.
0015The metabolic laboratory is specialized in the
investigation of body fluids in the search of abnor-
mal metabolites which are present in many inborn
errors of metabolism. Apart from the routine investi-
gations mentioned above, liquid, gas, and thin-layer
chromatography are performed on plasma, urine and
cerebrospinal fluid, and mass spectrometry, electro-
phoresis and spectrometric analysis techniques are
other methods available for investigation. The use
and distribution of these techniques in the investiga-
tion of patients suspected of an inborn error of
metabolism are depicted in Figure 1.
0016A systematic description of the metabolites and
abnormalities frequently observed in inborn errors
of metabolism is beyond the scope of this overview.
In practice, the inborn errors in which laboratory
abnormalities are found are grouped using three dif-
ferent conceptual approaches. Grouping takes place
by: (1) the main laboratory abnormality in a body
fluid (e.g., aminoacidemia or organic aciduria) as a
result of the enzyme defect; (2) the cell organelle
where the abnormality is located (e.g., lysosomal
storage disease, perixisomal disorder); and (3) the
abnormal structural molecule (e.g., defect of purine
metabolism, mucopolysacharidosis, or congenital
disorder of glycosylation). Figure 2 shows the distri-
bution of diagnosis of inborn error of metabolism
based on records in one laboratory of metabolic
diseases where both regional and international
patient investigations take place.
0017The diagnosis system of an inborn error of metab-
olism is eventually based on the enzyme nomenclat-
ure, the MIM number (Mendelian inheritance in
INBORN ERRORS OF METABOLISM/Overview 3265
man, formerly the McKusick number) for each dis-
ease entity, and the mutations linked with the clinical
entities in the OMIM database (Online Mendelian
inheritance in man: Table 1). Enzyme diagnosis,
using leukocytes, cultured fibroblasts, or biopsies
from chorionic villi or other affected tissue, is increas-
ingly used to confirm the diagnosis. DNA diagnosis is
also developed for an increasing number of inborn
errors.
0018 In the following paragraphs, some important dis-
orders and clinical problems are presented, to illus-
trate the interactions between nutrition and inborn
errors of metabolism.
Amino Acid Disorders (Group 2
Pathophysiology)
0019 Amino acids are the building blocks of body protein
and are mainly derived from the diet. Ingested
protein is absorbed almost completely. Amino acids
which become available in the body, in excess of the
needs for protein synthesis and growth, are eventu-
ally oxidized through several pathways. For the non-
essential amino acids there are also pathways of
biosynthesis. Some of these pathways are shared be-
tween two or more amino acids, and others are unique
to one amino acid. Inborn errors of metabolism are
known for each amino acid. Here we discuss two of
the most frequently encountered aminoacidemias.
Hyperphenylalaninemia
0020Phenylalanine is an essential amino acid. It is nor-
mally degraded via the tyrosine pathway. To enter
the tyrosine pathway, phenylalanine is converted
into tyrosine by the enzyme phenylalanine hydroxy-
lase, which has tetrahydrobiopterin as a cofactor.
Deficiency of the enzyme or of its cofactor causes
accumulation of phenylalanine in the body fluids
and tissues. Hyperphenylalaninemia is present, and
detection in plasma is a reliable way of establishing
the suspected diagnosis.
0021Classic phenylketonuria (PKU) is caused by the
deficiency of the enzyme phenylalanine hydroxylase.
In the first weeks after birth patients have no symp-
toms, although in the neonatal period vomiting can
be an early symptom. Mental retardation is the major
abnormality in untreated patients, with an estimated
loss of about 50 IQ points by the end of the first year
37%
2%
6%
8%
14%
1%
32%
Liquid chromatography
Thin-layer chromatography
Gas chromatography
Mass spectometry
Spectrometric analysis
Electrophoresis
Routine analyses
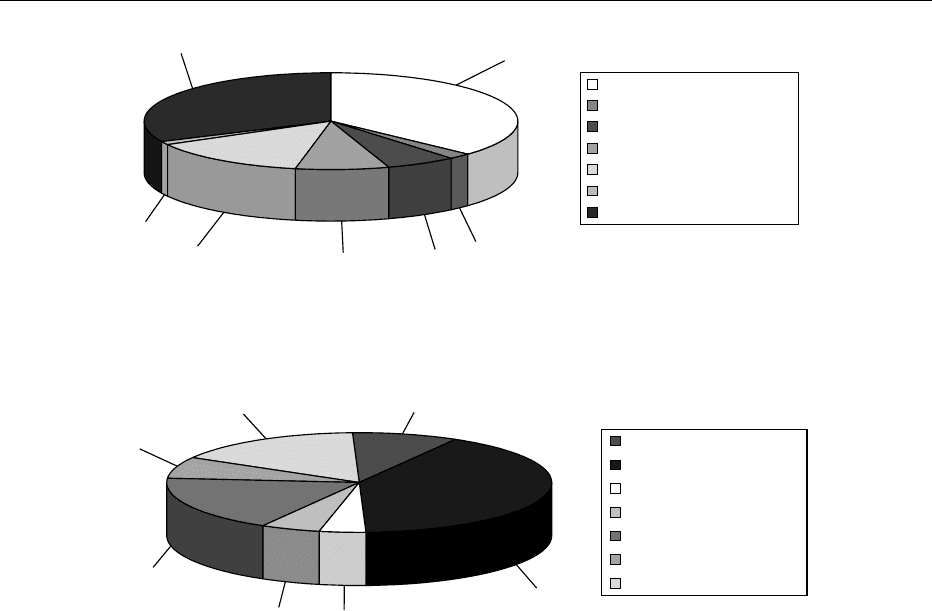
fig0001 Figure 1 (see color plate 99) Laboratory analyses performed in body fluids of 2150 patients investigated over a period of 5 years in
the laboratory for metabolic diseases of the Vrije Universiteit Medical Center in Amsterdam. Investigations were performed in whole
blood, plasma, urine, amniotic fluid, and cerebrospinal fluid.
18%
Aminoacidemias
Organic acidurias
Carbohydrate disorders
Lysosomal disorders
Peroxisomal disorders
Mitochondrial disorders
Rest
9%
41%
4%
5%
7%
16%
fig0002 Figure 2 (see color plate 100) Frequency distribution according to diagnostic group in 263 patients newly identified with an inborn
error of metabolism over a period of 5 years in the Laboratory for Metabolic Diseases of the Vrije Universiteit Medical Center in
Amsterdam. Fatty acid oxidation defects included in organic acidurias.
3266 INBORN ERRORS OF METABOLISM/Overview

of life. Untreated patients develop behavioral and
neurological abnormalities, including hypertonicity
and athetosis, and 25% of patients develop epilepsy.
On clinical examination they have an unpleasant
smell, probably due to the accumulation of abnormal
metabolites of phenylalanine such as phenylacetic
acid (which has a musky odor). Microcephaly
and growth retardation are common. If protein and
phenylalanine restriction is instituted early after
birth, the patients remain free of these symptom.
Special PKU infant formulas are available, but careful
follow-up of the patient’s physical growth and devel-
opment is needed and regular measurement of
phenylalanine plasma levels are mandatory in order
to prevent under- and overtreatment. Metabolic de-
rangement can take place under catabolic conditions,
e.g., during infections.
0022 Proper treatment requires early detection. Mass
newborn screening for PKU became effective when
Guthrie developed a bacterial inhibition assay which
could be done on dried blood collected on paper
obtained after a heel puncture during the second
half of the first week of life.
0023 About one in 50 patients with hyperphenylalanine-
mia have a defect in one of the enzymes necessary
for the synthesis or recycling of the cofactor tetrahy-
drobiopterin. If undetected and treated as PKU, these
patients show normalized phenylalanine plasma
levels but develop severe neurological symptoms.
The reason is that tetrahydrobiopterin is a cofactor
for other hydroxylases, involved not only in hydro-
xylation of phenylalanine but also of tyrosine and
tryptophan. The latter two are involved in the bio-
synthesis of the neurotransmitters dopamine and
serotinin. For this reason all neonates with hyperphe-
nylalaninemia are required to undergo tests for defi-
ciency of the cofactor. Treatment of these patients
requires protein restriction and supplementation
with tetrahydrobiopterin (because they also have
nonclassical PKU), and supplementation of l-dopa
and 5-hydroxytryptophan (because the supplemented
cofactor does not cross the blood–brain barrier).
0024 Research has demonstrated that in vivo hydroxyla-
tion in children with PKU can be so low that patients
may suffer from the effects of a diminished availabil-
ity of the metabolic product of phenylalanine hydro-
xylase, tyrosine. Tyrosine is an essential amino acid
required for physical growth but also a precursor for
dopamine biosynthesis. Tyrosine supplementation
thus is required in some patients with PKU.
0025 PKU is one of the more common inborn errors of
metabolism and this autosomal recessive disease is a
major cause of preventable hereditary mental retard-
ation and neurological debilitation in the population.
Neonatal screening programs have saved thousands
of patients who would otherwise have spent their life
in asylums. Nowadays, many adult patients cease the
diet and report no psychological or neurological
problems. None the less, new problems have emerg-
ed in the management of these patients. Pregnancies
in mothers with PKU who no longer use the low-
phenylalanine diet are complicated by spontaneous
abortions, and their living offspring are often men-
tally retarded and show microcephaly or congenital
heart disease. It has become clear that women with
PKU who are of child-bearing age should start a low-
phenylalanine diet before conception and should be
closely monitored for their phenylalanine blood levels
before and during pregnancy. Also, deficiency of n-6
polyunsaturated fatty acids has been described in
patients with PKU (and supplementation has been
advocated). The complications during pregnancy
and of polyunsaturated fatty acid synthesis demon-
strate one of the concepts of group 2 pathophysiol-
ogy, i.e., that metabolites accumulating proximal to
the metabolic block can cause derangements in dis-
tant organs and metabolic pathways.
Tyrosinemia Type 1
0026Tyrosine is likewise an essential amino acid derived
from ingested protein. Excess tyrosine is oxidized.
Several inborn errors of metabolism in the degrada-
tive pathway are known. Deficiency of fumarylace-
toacetate hydroxylase causes type 1 tyrosinemia, and
has an acute or chronic clinical manifestation. The
acute form presents in infancy, and comprises most
reported cases. Some symptoms resemble that of PKU
with failure to thrive, developmental delay, and
vomiting, but hepatic manifestations with organ
enlargement, jaundice, and bleeding tendency are
common findings as well. Apart from elevated tyro-
sine, elevated methionine is also found in many pa-
tients. Hypoproteinema and low prothrombin are
often present. Many patients develop end-stage hep-
atic failure in childhood and die unless appropriate
therapy is started. Patients with the chronic form also
present with failure to thrive and development delay,
but generally not during the first year of life. Cirrhosis
(which can be associated with acute liver failure
during catabolic episodes), renal tubular dysfunction,
and vitamin D-resistant rickets are often found. Acute
episodes of polyneuropathy may complicate the dis-
ease. In the long term, a substantial proportion of
patients with type 1 tyrosinemia develop hepatic ad-
enoma, and over time hepatocellular carcinoma can
develop in these lesions. The wide spectrum of organ
pathology, including malignant disease in tyrosine-
mia, demonstrates the effect of toxic substances
that accumulate due to the inability to catabolize
tyrosine. Although dietary restriction of tyrosine and
INBORN ERRORS OF METABOLISM/Overview 3267

methionine has been advocated over the years, many
patients have shown progression of hepatorenal and
hematological symptoms and developed malignant
liver disease under this treatment regimen. Liver
transplantation is a cure for patients with liver cirrho-
sis or (solitary) tumors, as the donor liver provides the
patients with a normally functioning liver, including
normal activity of the deficient enzyme. Liver trans-
plantation has a high risk of acute and chronic
complications and thus is only an option for patients
with severe liver disease. Since 1994 a promising
new pharmacological therapy has been successful in
reducing morbidity and liver complications: nitro-
trifluoro-methylbenzoyl-cyclohexanedione (NTBC),
an inhibitor of p-hydroxyphenylpyruvate dioxygen-
ase, prevents the formation of (cytotoxic and carcino-
genic) metabolites distal of this enzyme and proximal
of the inborn error. Although the elevated tyrosine
levels do not disappear during NTBC therapy, and
tyrosine and methionine restriction remain necessary,
this treatment appears remarkably effective. Patient
follow-up of this treatment has been less than 10
years at present. There is hope that the long-term
life-threatening complications such as liver failure,
bleeding disorders and tumor induction are prevent-
able with NTBC.
Energy Disturbance (Group 3
Pathophysiology)
Hypoglycemia
0027 A low blood sugar level is potentially dangerous (for
central nervous system function) and can be life-
threatening. Some organs (the heart, the brain) have
a high energy expenditure while using only a limited
selection of metabolic fuels. For the brain the balance
between energy supply and expenditure is the most
critical because glucose and ketone bodies (and,
under certain conditions, lactate) are its only energy
substrates and the specific energy demand is high.
Energy stores in the neural cells are very low and
rapidly consumed. Although hypoglycemia can be
caused by nonmetabolic disorders it is a hallmark
symptom for a large number of inborn errors of
metabolism, particularly those of intermediate carbo-
hydrate and triacylglycerol metabolism (Table 3).
0028 Symptoms can be present within hours after birth
(and, in the case of low liver stores, even within
minutes). The time of presentation after the last
meal is indicative of the relevant group of disorders,
with glycogen storage disease, gluconeogenesis
defects, and fatty acid oxidation defects and impaired
availability of ketones successively being the most
probable cause when a wider gap is reported between
the time of the last meal and the presentation of the
hypoglycemia. For the diagnosis of the inborn error,
and for the differential diagnosis, including endocrine
disorders, blood and urine sampling at the time of the
hypoglycemic crisis is essential. In life-threatening
situations such as coma, treatment prevails over the
complete collection of samples for the diagnostic
work-up. Some abnormalities may still be present in
blood and urine even hours after intravenous admin-
istration of glucose and restoration of the blood
glucose level to the normal range. In some disorders
symptoms can develop only months or even years
after birth. The clinical spectrum is wide, with liver
enlargement present in some (e.g., glycogen storage
type I and III, hereditary fructose intolerance) but not
all disorders and toxic crises present in some (e.g.,
coma and severe liver function abnormalities in
medium-chain acyl-coenzyme A dehydrogenase defi-
ciency) but not all disorders. Hyperlipidemia, gran-
ulocyte dysfunction, hyperuricacidemia and gout,
hyperlactatemia and osteoporosis, liver adenomas,
and pancreatitis are complications in just one of
these disorders (glycogen storage disease type I). Cat-
aracts with or without liver dysfunction are seen in
galactosemia. This illustrates that virtually any organ
tbl0003Table 3 Metabolic causes of hypoglycemia in childhood
Decreased production of glucose
Decreased release of glucose from the liver
Glycogen synthase deficiency
Glucose-6-phosphate deficiency (GSD type Ia or Ib)
Amylo-1,6-glucosidase deficiency (GSD type III)
Galactose-1-phosphate deficiency (galactosemia)
Fructose-1-phosphate aldolase deficiency (hereditary fructose
intolerance)
Decreased rate of gluconeogenesis
Pyruvate carboxylase deficiency
Phosphoenolpyruvate carboxylase deficiency
Fructose-1,6-diphosphatase deficiency
Glycerokinase deficiency
Decreased availability of alternative fuels, resulting in increased use
or decreased conservation of glucose
Impaired oxidation of fatty acids
Medium-chain acyl-coenzyme A dehydrogenase deficiency
Long-chain acyl-coenzyme A dehydrogenase deficiency
Carnitine acyltransferase I deficiency
Multiple acyl coenzyme A dehydrogenase deficiency
Impaired synthesis or use of ketones
b-Ketothiolase deficiency
Hydroxymethylglutaryl coenzyme A lyase deficiency
Decreased fat stores
Prematurity
Malnutrition
GSD, glycogen storage disease.
Other metabolic disturbances resulting in hypoglycemia are toxic
(exposure to ethanol or salicylate), endocrine (hyperinsulinemia, growth
hormone deficiency, cortisol deficiency), hyperleucinemia, and a self-
limiting ketotic hypoglycemia.
3268 INBORN ERRORS OF METABOLISM/Overview

system can be affected in the wide spectrum of dis-
orders encountered as the cause of a decreased blood
level of just one metabolite, glucose.
0029 A systematic overview of the spectrum of disease
and complications of inborn errors of metabolism
causing hypoglycemia cannot be further pursued
here. Treatment depends on the cause, and ranges
from strict avoidance of certain foods (e.g., galactose
and lactose in galactosemia) to continuous drip-
feeding (e.g., in glycogen storage disease type I).
Avoidance of prolonged fasting is important in a
large proportion of disorders. Early diagnosis is es-
sential to prevent brain damage and other irreversible
organ failure. Thus recurrent or severe hypoglycemia
should bear a high degree of suspicion of a metabolic
cause.
Elevated Resting Energy Expenditure
0030 Mitochondrial respiratory chain enzyme defects are
associated with myopathy and failure to thrive in
early life in mildly affected patients and severe multi-
organ involvement, including brain damage and early
death in other patients. The failure to thrive in infants
with the disease can result from neurological impair-
ment (vomiting, difficulty with swallowing) but also
elevated energy expenditure is often found. The
pathophysiology of the increased resting energy ex-
penditure is of conceptual interest for this overview
on human inborn errors and nutrition. The mitochon-
drial respiratory chain is comprised of five enzyme
complexes which together drive the process of oxida-
tive phosphorylation and maintain the proton gradi-
ent through which adenosine triphosphate (ATP)
production is maintained. Complex I, III, and IV
have proton-pumping capacity, and the efficiency of
the protons (with eventually energy transfer to ATP,
P) pumped and coupled electron flow through the
respiratory chain to oxygen (O) can be expressed as
the P/O ratio. In normal mitochondria the ratio is
about 1.75 for reduced nicotinamide adenine
dinucleotide (NADH)-linked substrates and 2.75 for
reduced flavin adenine dinucleolide (FADH)-linked
substrates. However, in patients with deficiency of
complex I, II, or III, the respiratory chain is substan-
tially less efficient as one proton (ATP) less is
produced by the electron’s energy transfer between
complex I and V. Adaptive changes, including a
higher adenosine diphosphate (ADP) concentration
(a known stimulus of oxidative phosphorylation
activity) and higher resting oxygen consumption are
present in many patients. To find out whether, and to
what extent, this pathophysiological mechanism is
present in an individual patient diagnosed with a
respiratory chain defect indirect calorimetry can be
useful. Therapy exists in high-energy feedings, and
growth monitoring is important. This is particularly
important during infancy when energy demands and
growth rates are already high and patients depend on
food offered by the mother. In later life many patients
expend less energy due to muscle fatigue and lower
physical activity levels, and the balance between total
daily energy expenditure and energy intake is more
easily maintained.
Lipids and Essential Fatty Acid Deficiency
0031In the affluent world and in poor parts of the world,
nonhereditary causes of insufficient essential fatty
acid status prevail and are worldwide much more
frequent than hereditary causes. Disturbance of n-6
and n-3 polyunsaturated fatty acid status is caused
by a number of inborn errors. The most frequent ones
are shown in Table 4. Single-enzyme deficiencies in
the metabolic pathways of n-6 and n-3 polyunsatur-
ated fatty acid synthesis and oxidation are extremely
rare, probably because essential fatty acids from food
and the interconnections between the n-6 and n-3
desaturase pathways provide alternative provisions
and overflow routes for affected metabolites. These
rare patients are not reviewed here or included in
Table 4.
tbl0004Table 4 Causes of essential fatty acid deficiency
Mechanism and disease state Affected gene(s)
Hereditary causes
Intestinal malabsorption
Abetalipoproteinemia MTTP
Hypobetalipoproteinemia Apolipoprotein B
Anderson disease ?
Exocrine pancreas insufficiency
Cystic fibrosis CFTR
Shwachman syndrome Mapped to 7p11-q11
Pearson syndrome Mitochondrial DNA deletions
Deranged metabolism of
polyunsaturated fatty acids
Decreased synthesis
of docosahexanoic acid
Phenylketonuria PAH
Zellweger syndrome PEX
Decreased inhibition of
leukotriene B
4
Sjo
¨
gren–Larsson syndrome FALDH
Nonhereditary causes None
Malnutrition
Fat-free diet
Parenteral nutrition without
essential fatty acid
supplementation
MTTP, microsomal triglyceride transfer protein; CFTR, cystic fibrosis
transmembrane conductance regulator; PAH, phenylalanine hydroxylase;
PEX, peroxin, involved in peroxisome biogenesis; FALDH, microsomal
fatty aldehyde dehydrogenase.
INBORN ERRORS OF METABOLISM/Overview 3269