Marcus P. Corrosion mechanisms in theory and practice
Подождите немного. Документ загружается.

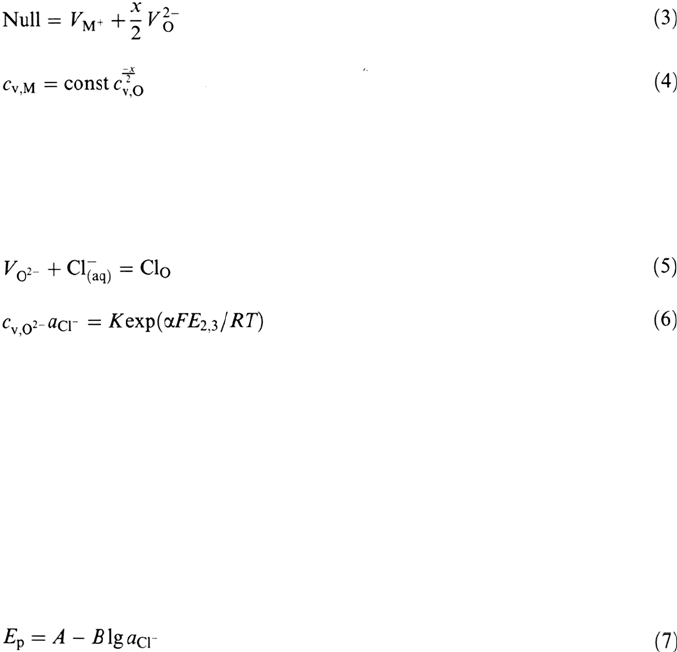
On the basis of these ideas, Macdonald and co-workers [13,14] developed
their model of passivity and its breakdown involving the action of vacancies within
the passive layer. It is assumed that cation vacancies migrate from the oxide-
electrolyte to the metal-oxide interface, which is equivalent to the transport of
cations in the opposite direction. If these vacancies penetrate into the metal phase
at a slower rate than their transport through the oxide, they accumulate at the
metal-oxide interface and finally lead to a local concentration. The related voids
lead to stresses within the passive film and its final breakdown. The inward diffusion
or migration of cation vacancies is affected by the incorporation of Cl ions at the
oxide-electrolyte interface according to the following mechanism: The concentration
c of metal ion V
M+
, and O
2–
vacancies V
O
2–
are determined by the equilibrium of
the Schottky pair formation at the oxide-electrolyte interface [Eq. (3)], which
causes an inverse dependence of their concentrations [Eq. (4)].
Mechanisms of Pitting Corrosion 249
In the presence of Cl
–
, its incorporation in O
2–
vacancies occurs according to the
equilibrium of Eq. (5), which is affected by the potential drop E
2,3
at the interface
according to Eq. (6) with the concentration of oxygen vacancies c
v,O
2–
at the surface,
the activity of chloride a
Cl
–
within the solution, and the potential-independent part
of the equilibrium constant K.
According to Eq. (6), c
O
2–
depends on a
Cl
–
and the potential drop E
2,3
; c
O
2–
in turn
affects c
v,M
, which is the driving force of the diffusion of the cation vacancies to
the metal-oxide interface. Thus the interdependence of the concentrations of
cation and anion vacancies within the oxide and the incorporation of Cl
–
determine
the concentration gradient of cation vacancies and their transport through the
oxide layer that will cause a critical concentration for breakdown at the pitting
potential. According to this outline, the further discussion yields a semilogarithmic
dependence of the pitting potential E
p
and the chloride activity a
Cl
–
within the
electrolyte similar to Eq. (1) [Eq. (7)]. The constant B contains the diffusivity
constant of the cation vacancies within the oxide layer.
Objections to the point defect model are that in its original form it assumes linear
transport equations with diffusion of the different species within the passive layer,
whereas migration with an exponential dependence on the high electrical field
strength of some 10
6
V/cm should be dominating as a driving force. It also
concentrates the changes in electrode potential ΔE to the potential drop E
2,3
at the
oxide-electrolyte interface, so that it fully enters the equilibrium of Cl
–
incorporation
of Eqs. (5) and (6). However, large parts of ΔE contribute to Δφ, i.e., are located within
the barrier part of the passive layer (Fig. 2), and E
2,3
changes only with the
Copyright © 2002 Marcel Dekker, Inc.
composition of the oxide surface, if at all, and the pH of the solution but not with
the applied electrode potential, at least for stationary conditions. An overpotential η
2,3
for the potential drop E
2,3
is mainly expected for nonstationary conditions of the passive
layer that will directly influence the adsorption equilibrium of Cl
–
. The specific role of
chloride and other halides in breakdown of passivity is still not sufficiently understood
in light of this theory. Further refinements might improve this interesting view.
Surface analytical methods unfortunately do not always give a clear answer
about the penetration of aggressive anions. Some authors found chloride within the
film with XPS, Auger electron spectroscopy (AES) [15,16], and secondary ion mass
spectroscopy (SIMS) [17]; others could not find it within the film [18–22]. The
contradictory results may be explained in terms of sample preparation and the
sensitivity of the methods. Very careful XPS studies with Fe-Cr alloys show
incorporation within the outer hydroxide part of the duplex passivating film [23,24].
The inner oxide layer remains free of Cl
–
if prepared within chloride-free electrolytes
before exposure to the aggressive anions. Similarly, incorporation of Cl
–
within the
outer hydroxide layer was found for pure Ni [25]. It was found in the inner oxide part
only when the passive layer was formed in solutions already containing Cl
–
. If the
electrode potential was above the critical value for breakdown, Cl
–
penetrated
into the preexisting oxide with possible lateral fluctuations of its concentration,
leading finally to the formation of pits. According to these studies, the accumulation
within the hydroxide overlayer serves as an accumulation of a sufficiently large
amount to cause breakdown in the following step. Further discussions of the
surface analytical investigations of passive layers that have been exposed to
chloride-containing solutions may be found in the chapter on passivity in this book.
A detailed bilayer or even multilayer structure is observed for passive films on
many metals and alloys [26,27]. The outer part is usually a hydroxide, whereas the
main inner part is an oxide [23,25–27]. The hydroxide structure may well act as an
ion exchanger or at least absorb anions, as has been proved for some systems.
Although the access of aggressive anions leads to changes of the passive layer
detected by ellipsometry [28] and reflection spectroscopy [29], it is still unclear what
conclusion may be drawn from these observations. If the penetration of aggressive
anions leads to weak channels where intense dissolution may start, it is unclear why
the film does not re-form specifically at this site and why these defects do not
repassivate. The self-healing mechanism of the passive layer is essential for its
excellent protecting property. The specific role of the aggressive anions is missing
in this mechanism of breakdown. Any explanation should involve the characteristic
chemical properties of anions such as the halides.
Experiments with well-prepassivated specimens show that the formation of a
corrosion pit may be an extremely fast process, in the time range of <1 s or even
<1 ms [4,30]. Figure 3 depicts as an example the increase of the current density within
less than 1 ms as a consequence of growing pits for an Ni specimen prepassivated
for 1 s before a potential shift above the critical pitting potential. A simple
comparison of the small stationary passive current densities in the range of μA/cm
2
with these short times leads to contradictions with the penetration mechanism
[30,31]. If anions migrate inward as cations migrate outward during stationary
dissolution, these fast nucleation times cannot be understood. Furthermore, it seems
questionable that large anions such as SO
4
2–
and ClO
4
–
migrate sufficiently fast in the
250 Strehblow
Copyright © 2002 Marcel Dekker, Inc.
electrical field through the passive layer to cause nucleation in times t < 1 s
[30,31]. These anions may also cause pitting of iron at the lower potential part of
the passivity range and ClO
4
also at potentials close to the transpassive range
[2,3]. However, the inward migration of aggressive anions and the outward
migration of cations may be facilitated at local defects within the passive layer.
Even for those situations, one needs the special chemical properties of the
anions to understand why the defects do not repassivate but develop to a corro-
sion pit. Any mechanism of nucleation and growth of corrosion pits has to
include the specific role of the aggressive anions that causes the formation of a
pit instead of repassivating the defect site. These details will be discussed in one
of the following sections.
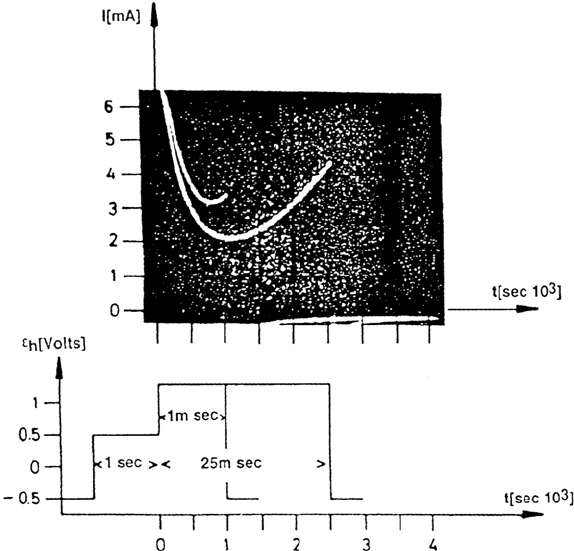
In this regard, one experiment with well-passivated Fe electrodes should be
mentioned [6]. If the penetration of aggressive anions through the passive layer was
the rate-determining step, a high electrical field strength should favor their migration
and accelerate pit nucleation. For nonstationary conditions, for passive Fe, exactly
the contrary has been observed (Fig. 4). After passivation for 1 h at 1.18 V (SHE) in
phthalate buffer pH 5.0 plus 0.1 M K
2
SO
4
, the nucleation rate was severely increased
when the potential was stepped to 0.78 V immediately before Cl
–
addition to 0.01 M.
This pretreatment resulted in a much faster increase of the current density with time
and a slope of 3 for a double logarithmic current-time plot was found, which is an
indication of an increased nucleation rate that is constant with time. These
Mechanisms of Pitting Corrosion 251
Figure 3 Pit nucleation of Ni (A = 0.02cm
2
) at E = 1.30 V (SHE) in phthalate buffer, pH
5.0 + 0.1 M KCl after 1 s prepassivation at E = 0.5 V. (From Ref. 30.)
Copyright © 2002 Marcel Dekker, Inc.
observations were confirmed by direct microscopic examination of the specimen’s
surfaces. If one waits at the lower potential before chloride addition, the nucleation
is reduced again with a related change of the current increase from a slope 3 to 2
for the double logarithmic plot, which indicates a variation from a constant
nucleation rate to a constant number of pits, i.e., no further nucleation. Apparently
the nucleation is severely increased for nonstationary conditions and is reduced
again when approaching a new stationary state. An explanation of this effect with
the penetration mechanism leads to contradictions. When the potential is stepped
to lower values the electrical field strength is decreased and one expects reduced
rather than increased migration of the aggressive anions through the passive layer
(Fig. 2). Remaining at the lower potential before Cl
–
addition should decrease the
thickness of the passive layer because of an excess of film dissolution over its
formation. The approach to the new stationary state with time at a lower potential
252 Strehblow
Figure 4 Double logarithmic plot of the increase of the geometric current density with time
for electropolished iron during pitting corrosion, 1 h prepassivated at 1.18 V and potential drop
to 0.78 V (solid line), Δt = time between potential change and chloride addition, 1 h prepassivated
at 0.78 V (dashed line), phthalate buffer, pH 4.9, 0.1 M SO
4
2–
, 0.01 M Cl
–
. (From Ref. 6.)
Copyright © 2002 Marcel Dekker, Inc.
should yield the original field strength with an increase of penetration. Again, the
opposite effect was found [6]. Even the adsorption mechanism cannot explain
these observations. If the potential is decreased, the voltage drop E
2,3
at the oxide-
electrolyte interface will decrease intermediately with a negative overvoltage η
2,3
for O
2–
formation from the water, i.e., the dissolution of the oxide, until a new
stationary state with E
2,3
= E
2,3,s
is achieved (Fig. 2). This situation should disfavor
the adsorption of Cl
–
at this interface, which again should slow down the penetration
instead of increasing it.
Film Breaking Mechanism
The occurrence of fissures within the passive layer is a possible explanation for the
observations mentioned last, especially for an nonstationary state of the passive
layer. A sudden change of the electrode potential even in a negative direction will
cause stresses within the film. Chemical changes [32] or electrostriction [7,8] is a
reasonable explanation. Chemical changes, i.e., a reduction of Fe(III) to Fe(II),
have been detected with XPS when negative potentials were applied to electrodes
passivated at positive potentials well within the passive range [32]. There also
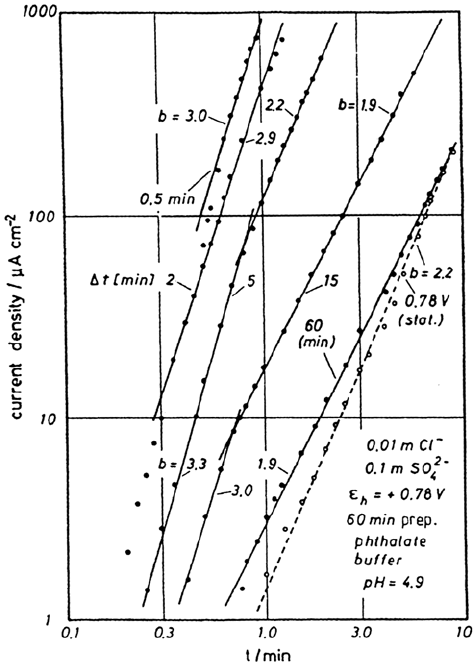
exists direct evidence of film breaking events for these nonstationary conditions
according to observations with a rotating Fe-disk-Pt-ring electrode [33]. Even in
the absence of aggressive anions, a sudden potential decrease from 1.3 to 0.7 V
causes release of Fe
2+
ions after a few seconds into the electrolyte, as detected by
a temporary peak of the analytical ring current detecting Fe
2+
ions (Fig. 5). This is
a consequence of film breaking and repair events. Any crack within the passive
layer will lead to direct contact of small parts of the metal surface with the electrolyte
and thus to dissolution of Fe as Fe
2+
with current densities that refer to the relatively
positive potentials, but without any protecting oxide at these defect sites. The
self-healing mechanism of the passive layer causes only temporary Fe
2+
formation
of a few seconds duration. These potential changes should cause numerous defects
of nanometer dimensions within the passive layer in order to get a sufficient
amount of unprotected metal surface so that the temporary release of Fe
2+
becomes
measurable. In the presence of aggressive anions, their direct access to the metal
surface will prevent repassivation and pits can form. The serious increase of the
nucleation rate (Fig. 4) after these potential changes is further evidence for the
suggested formation of a large number of defects in the nanometer range.
As already mentioned, pit nucleation is a very fast process. For well-passivated
Fe specimens, the time for the occurrence of a first pit is less than a second for
potentials well above the critical value E
p
, as determined by the increase of the
current density after the addition of chloride to the solution and direct microscopic
observation with a camera [4]. Most of the time is used to get the aggressive anions
to the specimen surface by convection. If chloride is already present in the electrolyte,
a short passivation below the pitting potential for approximately 1 s and a subsequent
potential increase cause pit formation within less than a millisecond [4]. Results
obtained with these electrochemical pulse techniques suggest that nonstationary
conditions of the passive layer favor the film breaking mechanism. The penetration
of aggressive anions would require much longer times than observed during these
pulse experiments. If the experimental conditions are not in favor of pitting, i.e., for
Mechanisms of Pitting Corrosion 253
Copyright © 2002 Marcel Dekker, Inc.
small concentrations of aggressive anions (<10
–3
M) or low potentials in the vicinity
of E
p
, pit nucleation requires times longer than these extremely short times.
Long passivation times suggest that film breaking is a good explanation for
the pit nucleation for nonstationary conditions too. The current increase caused by
pitting slows down when the passivation time is increased up to more than 10 h
[6,31]. The passive layer is formed during the first seconds and does not grow
much after that time. This is a direct consequence of the barrier character of these
films, which leads to logarithmic or inverse logarithmic growth with time. Further
decrease of the passive current density and especially of the pit nucleation after
hours may easily be explained by constantly occurring film breaking and repair
events. The related stresses will heal with passivation time so that these events
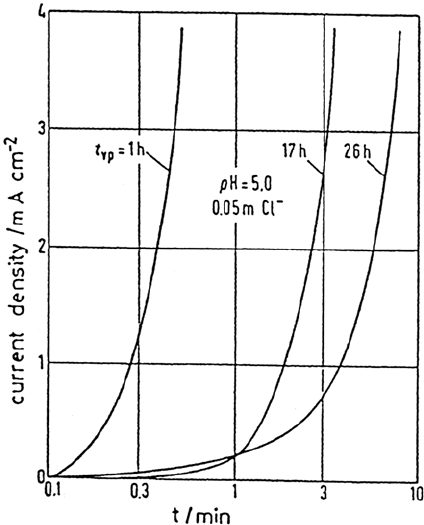
become rare with time. Figure 6 shows an example of a decrease of pitting with
passivation time and the related reduction of the current increase [31].
254 Strehblow
Figure 5 Disk current I
D
and ring currents I
R1
for Fe
3+
analysis and I
R2
for Fe
2+
analysis
for a potential variation from E
D
= 0.7 V to 1.30 V and back. E
R1
= 0.10 V, E
R2
= 1.40 V,
0.5 M H
2
SO
4
, pH 0.3, f = 1000 min
–1
, disk area A = 0.09cm
2
. (From Ref. 33.)
Copyright © 2002 Marcel Dekker, Inc.
Measurement of the electrochemical current noise has the aim of correlating
the observed current fluctuations with breakdown and repair events that might lead
to the formation of stably growing pits [34,35]. In the view of these theories, the
application of statistical methods to the occurrence of current spikes and the
observed probability of pit formation lead to a stochastic model for pit nucleation.
The evaluation of current spikes in the time and frequency domains yields parameters
such as the intensity of the stochastic process λ and the repassivation rate τ [34].
They depend on parameters such as the potential, state of the passive layer, and
concentration of aggressive anions.
An interesting discussion of current measurements on microdimensional
electrodes of stainless steel wires is given by Mattin and Burstein [36]. Their
analysis of current transients at a very low level in chloride-containing 0.075 M
HClO
4
leads to the distinction of metastable and stable pits. According to their
discussion, the remaining passive layer protects the pit analyte from being diluted
from the bulk solution. Only when the film breaks off too small pits will repassivate,
whereas a few larger ones are deep enough to keep their local environment
undiluted so that they survive.
Adsorption Mechanism
The passive current density is influenced by the action of anions even for conditions
where pitting does not occur. Passive iron in 0.5 M H
2
SO
4
shows stationary current
Mechanisms of Pitting Corrosion 255
Figure 6 Increase of current density for passive Fe with pitting corrosion in phthalate buffer,
pH 5.0 and 0.05 M Cl
–
at E = 1.38 V for different prepassivation times t
pp
. (From Ref. 31.)
Copyright © 2002 Marcel Dekker, Inc.
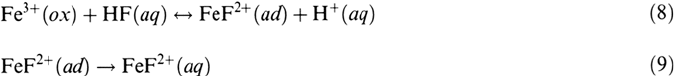
densities (7 μA/cm
2
) that are higher by approximately one order of magnitude
than for 1 M HClO
4
(<1 μA/cm
2
) [37]. This observation has been explained by a
catalytic effect of SO
4
2–
anions on the transfer of Fe
3+
from the oxide to the electrolyte
[38]. This cation transfer is the rate-determining step for the passive corrosion
reaction. In the presence of SO
4
2–
, an FeSO
+
4
complex forms. It is reasonable that
this complex, with only one positive charge, requires less activation energy to be
transferred from its O
2–
ligands within the oxide matrix to the electrolyte than the
highly charged Fe
3+
ion. The complexation of cations by organic reagents cause a
similarly enhanced dissolution in the passive state. The dissolution of Ni
2+
from
passive nickel and nickel base alloys is enhanced by organic acids such as formic
acid, which may lead to the removal of NiO in the passive layer [39]. Similarly,
additions of citrate to the electrolyte cause thinning of the passive layer on stainless
steel and increase of the Cr content within the oxide layer. This is a consequence
of accelerated transfer of Fe
3+
from the film surface to the electrolyte [40].
Apparently the complexation and transfer of Cr
3+
are not enhanced.
It is well established that metals that show passivity within strongly acidic
electrolytes are far from the dissolution equilibrium of the oxide. The barrier
character of the passive layers for these conditions requires slow dissolution kinetics
at the oxide-electrolyte interface. Similarly to the catalytic effect of SO
4
2–
and
complexing organic agents, halides enhance the transfer of Fe
3+
and Ni
2+
from the
oxide to the electrolyte. The adsorption mechanism for pit nucleation starts with the
formation of surface complexes that are transferred to the electrolyte much faster than
uncomplexed Fe
2+
ions (Fig. 2c). These details have been studied for Fe in Cl
–
- and
F
–
-containing solutions [21,41–43]. Similarly, Ni in solutions containing F
–
has been
studied [22,42–44]. Fe is a good metal with which to analyze the processes leading
to passivity breakdown because of the characteristic changes of the valence of
the dissolving ions, which may be measured with the rotating-ring-disk (RRD)
technique. Fe
3+
ions are dissolved if the metal surface is still covered completely with
Fe(III) oxide, whereas Fe
2+
is detected when bare metal comes into contact with
the electrolyte at a pit surface or at a defect site, similar to Fe dissolution in the active
state. The release of Fe
3+
ions immediately after Cl
–
has access to a prepassivated Fe
electrode was taken as a measure of locally enhanced dissolution of the oxide film.
This leads to a local thinning of the passive layer and finally to its complete
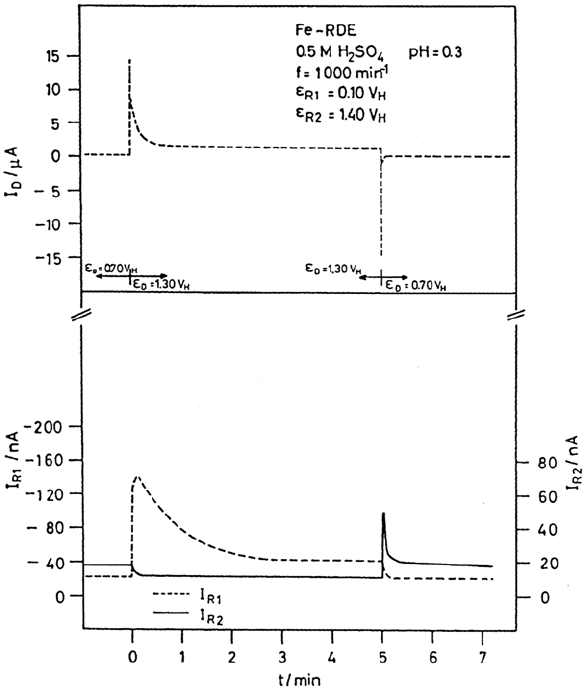
breakdown and the formation of a pit. In the case of F
–
in acidic electrolytes the attack
is more general; i.e., the passive layer is subject to a general attack. As a consequence,
the measured passive current density is increased by orders of magnitude (Fig. 7)
[21,42,43]. Unfortunately, the strong FeF
5
H
2
O
2–
complex that finally forms in
solution prevents its detection by its reduction to Fe
2+
at the analytical ring. The
current increases during the intermediate stage of attack with the first order of the HF
concentration. Therefore a proposed mechanism should yield an electrochemical
reaction order of one for HF. The following sequence of reactions has been proposed
for the processes that lead to thinning of the passive layer and enhanced passive
dissolution [21,42–44]:
256 Strehblow
Copyright © 2002 Marcel Dekker, Inc.

The adsorption (8) is assumed to be fast and in a quasi-equilibrium. Reaction (9) is
the rate-determining step, and (10) is a fast following reaction step in solution
leading to the stable fluoro-aquo complex. These assumptions lead to a first-order
process for HF according to the rate equations (15) and (16). The Butler-Volmer
equation (11) will hold for the rate-determining step (9) with a charge transfer
coefficient α and with the potential drop E
2,3,s
at the oxide-electrolyte interface for
stationary conditions. E
0
2,3,s
refers to a solution of pH 0. According to the equilibrium
of O
2–
formation of Figure 2a and Eq. (13), one obtains Eq. (14) for the pH
dependence of the potential drop E
2,3,s
for stationary conditions of the passive layer:
Mechanisms of Pitting Corrosion 257
Figure 7 Current-time dependence of a rotating Pt-split-ring-Fe-disk electrode in 1 M
HClO
4
after HF addition to 0.1 M, 2 h prepassivation at 0.1 V in 1 M HClO
4
, Fe
2+
detection
at ring 1. (From Ref. 21.)
Introducing equilibrium reaction (8) with its constant K according to Eq. (12):
and the equilibrium of O
2–
formation and the related pH dependence of the poten-
tial drop E
2,3,s
yields:
Copyright © 2002 Marcel Dekker, Inc.

The effect of HF is a model for pitting insofar as it involves the total passive
surface so that the measured effects are much more pronounced and refer to a surface
of known size instead of an unknown actual pit surface that even changes with time.
The later stages of the attack of the passive layer lead to current peaks that go along
with equivalent Fe
2+
formation (Fig. 7). Finally, a general breakdown of the passive
layer is observed with a steep increase of the dissolution current density and Fe
2+
for-
mation [21,42,43]. Apparently, one may observe the different steps of breakdown of
passivity for fluoride directly, which are difficult to follow for the local events in the
case of the other halides. The difference in the action of fluoride and the other halides
may be explained by a comparison of the stability constants of their iron complexes as
presented in Table 1 [30]. The stability constants K
1
with one anion are given, which
refers to the reaction order one that has usually been found for the disssolution of
passive layers under the influence of the aggressive anions. The reaction of Fe
3+
with
HF to FeF
2+
and H
+
yields the more realistic value of log K
1
= 2.28 due to the small
dissociation constant of HF (pK = –log K
d
= 2.98). Table 1 also contains the constants
K
1
for Ni
2+
- and Cr
3+
- halide complexes. Their falling values from fluoride to iodide
and Fe
2+
to Ni
2+
support the decreasing tendency for enhanced dissolution of the
passive layer and localized corrosion. These data can be referred to the situation at
the oxide surface. The fluoro complexes are very stable and form with high concen-
trations at all surface sites. Therefore their much faster transfer to the electrolyte
yields enhanced general dissolution, whereas the attack of the other halides is locally
restricted and much less pronounced. Besides thethermodynamically based values, i.e.,
the stability constants, the kinetics of complex formation and of the complex transfer
to the electrolyte are another decisive factor for the attack of the passive layer. In this
sense, the situation of Cr
3+
is very special and will be discussed separately.
XPS measurements of passivated Fe and Ni electrodes that have been
exposed to aggressive anions (Ni and Fe to F
–
; Fe to Cl
–
, Br
–
, and I
–
) but have
not already formed corrosion pits support this mechanism. The quantitative
evaluation of the data clearly shows a decrease of the oxide thickness with time
of exposure [22,48]. Not only F
–
but also the other halides cause thinning of the
passive layer (Fig. 8) [48]. The catalytically enhanced transfer of cations from
the oxide to the electrolyte leads to a new stationary state of the passive layer.
Its smaller thickness yields an increased electrical field strength for the same
potentiostatically fixed potential drop, which in turn causes faster migration of
the cations through the layer to compensate for the faster passive corrosion
reaction (1) at the oxide-electrolyte interface (Fig. 2a). Statistical local changes
258 Strehblow
Table 1 pK Values of Stability Constants of Metal Ion Complexes with Halides for the
Reactions Me
z+
+ X
–
→ MeX
(z–1)+
including Constants Referring to HF (*) in Acidic
Solutions [45–47]
Copyright © 2002 Marcel Dekker, Inc.