Marcus P. Corrosion mechanisms in theory and practice
Подождите немного. Документ загружается.

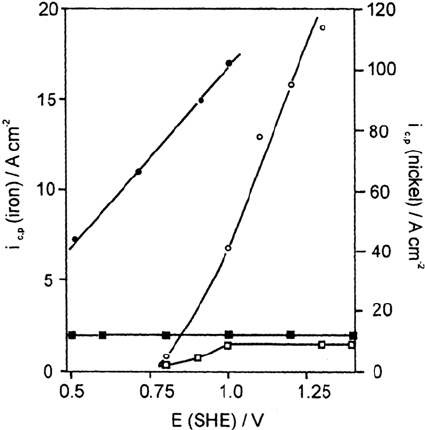
for Fe in 0.01 M KCl + 0.1 M KSO
4
(Fig. 15) [53]. Apparently the potential-
independent plateau values of i
c,p
are related to the formation of a salt layer that
provides electropolishing conditions at the pit surface (Fig. 13a). If, however, the
current becomes still smaller such as for Ni below 1.0 V, the growth of the pit
becomes unstable and irregular shapes are observed as shown in Figure 14.
A special geometry for localized corrosion studies has been applied by
Frankel and co-workers [59,60]. They subjected vapor-deposited thin Ni-20% Fe and
Al films on an inert substrate as working electrodes of a potentiostatic circuit to the
local attack of chlorides, which causes fast perforation of the film and subsequent
growth of two-dimensional pits with actively corroding cylindrical side walls. With
this method the authors avoid the usual deepening of three-dimensional pits on bulk
materials with the related increasing diffusion length for the transport of corrosion
products and ohmic drops. Thus, the geometry for metal dissolution and transport of
corrosion products was kept constant with time of growth of the two-dimensional
pits. For this simpler and constant geometry, the local current density and the
potential of repassivation of pits could be measured more accurately. The local
current densities i
c,p
could be directly deduced from the growth of the radius of the
two-dimensional pits applying Faraday’s law without any assumptions for the
geometry of the actively corroding surface. Usually, i
c,p
increases linearly with the
electrode potential up to 100 A/cm
2
, where it is controlled by mass transport and
ohmic resistance. It finally gets to a diffusion-controlled potential-independent
limiting value. There exist a critical local current density i
cr
and a repassivation
potential E
R
that are closely related to each other. The accumulation of corrosion
Mechanisms of Pitting Corrosion 269
Figure 15 Local pit current density i
c,p
, as a function of potential E deduced from pit
growth on polycrystalline nickel and Fe electrodes in 0.05 M phthalate buffer, pH 5.0. Ni:
() 0.1 M KCl + 0.1 M K
2
SO
4
, (c) 0.1 M KCl; Fe: () 0.01 M KCl + 0.1 M K
2
SO
4
, (z)
0.01 M KCl. (From Ref. 58.)
Copyright © 2002 Marcel Dekker, Inc.

products is interpreted by the authors as a stabilizing factor for pit growth. If the
local current density goes below a critical value i
cr
, localized corrosion will stop due
to a less aggressive environment corresponding to a smaller local concentration of the
corrosion products that may be maintained. Apparently, a sufficiently high concen-
tration of chloride is required to prevent repassivation. Estimates have shown that
~ 50% of the concentration of saturation is required to prevent repassivation [59]. As will
be shown later for a hemispherical pit, the locally increased concentration of corrosion
products and thus chlorides is proportional to the product of the local current density
and the pit radius i
c,p
r [Eq. (18)]. For a two-dimensional circular pit this relation
simplifies to a proportionality to i
c,p
which yields critical conditions that are inde-
pendent of the radius of the two-dimensional pit [59]. In this sense, these studies may
give valuable information on the stabilty of polygonal and hemispherical pits. With
increasing metal film thickness and consequently larger side walls of the actively
corroding cylinder surface, i
cr
decreases due to a larger corroding surface and hence
to an increased ohmic resistance and higher accumulation of corrosion products. For
similar reasons, di/dE decreases with increasing film thickness.
As already discussed in relation to the adsorption mechanism of pit nucleation,
the halides cause catalytically enhanced dissolution of the passivating oxide. Any
attempt of the bare metal surface of a pit to passivate by oxide formation will fail in
the presence of high concentrations of aggressive anions, similar to the galvanostatic
transient behavior of preactivated specimens. Thus, there is a close correspondence
of small iron and nickel electrodes in solutions of high Cl
–
content (>1 M) and an
intensively corroding pit surface that will be discussed in the next section. In both
cases, the formation of a passive layer is suppressed. In the case of Al a sufficiently
low pH by hydrolysis of the corrosion products will stabilize pitting additionally.
Passivity of this metal requires a dissolution equilibrium of the anodic oxide with the
electrolyte that requires weakly acidic to alkaline solutions. Localized acidification is
therefore a good additional stabilizing factor for corrosion pits on Al. This special
situation, however, does not contradict the necessary active chemical role of halides,
i.e., their complexing properties.
Precipitation of Salt Films
After a sufficiently long time of high dissolution current densities, the accumulation
of corrosion products will lead to the precipitation of a salt film. This precipitation is
expected in an early stage of pit growth if sufficiently large potentials and as a
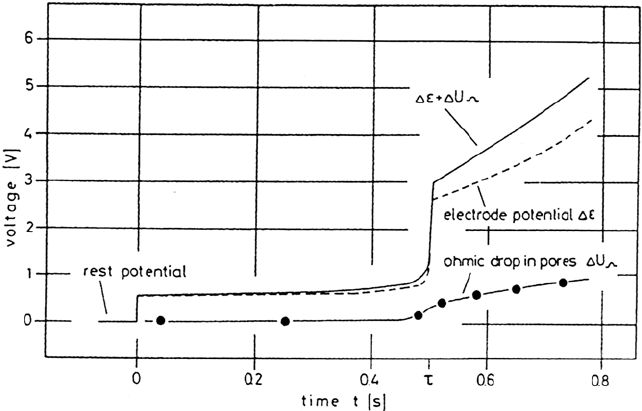
consequence large current densities are applied. Galvanostatic transients of
preactivated small electrodes in solutions of high chloride content are not subject
to passivation, and the dissolution at potentials within the passive range leads to a
related steep increase of the measured electrode potential after the transition time τ
(Fig. 16). For the linear diffusion problem of the completely dissolving electrode
surface it was found that Sand’s equation is valid; this relates the current density i
c,p
and time t of corrosion to the difference Δc = c
m
– c
m,b
of the metal ion concentration
with c
m
at the electrode surface and its bulk value c
m,b
, their charge z
m
, and diffusion
constant D of the metal ions [56].
270 Strehblow
Copyright © 2002 Marcel Dekker, Inc.
In the absence of supporting electrolyte, i.e., in a solution containing the salt
of the dissolving metal, a diffusion constant D
eff
= D(1 + z
m
/ | z
a
|), which includes
the contribution of migration to the transport, has to be used instead D. Here z
m
and z
a
are the valences of the metal ions and anions. Figure 17 presents the
accumulation of metal ions Δc according to Eq. (17) during dissolution of a small
nickel electrode in solutions of high chloride content as a function of i√t
–
. When
c
m
reaches its value of saturation c
m,s
for t = τ, precipitation of a salt film will
occur. This salt film has been examined further with galvanostatic pulses
superimposed on the main transient to determine ohmic drops (Fig. 16). The
results lead to a model for the forming salt film with a spongy, porous outer part
and a barrier-type inner part [56]. Besides experimental examinations [56], numerical
calculations [61] have shown that Eq. (17) may also be applied for concentrated
electrolytes. Different solutions with and without aggressive anions have been
tested using galvanostatic transients with small Fe and Ni electrodes, such as 0.5
M H
2
SO
4
, 1 M HClO
4
, 1 M NaClO
4
, and 1M HNO
3
. Figure 18 gives an example
for Fe. In the absence of aggressive anions, no potential plateau but only a small
shoulder was found for galvanostatic transients; i.e., only passivation with no
large free metal dissolution was observed. The product i√
τ
—
starts at small values
and decreases with increasing current density i. Only in the presence of > 1 M Cl
–
(or possibly other halides) were much larger values independent of i found for
i√
τ
—
. These studies also prove that the presence of aggressive anions is necessary
to prevent passivation of Fe and Ni surfaces.
For a hemispherical pit the diffusion-limited local current density i
c,p
changes
according to Eq. (18), which is a simple combination of Fick’s first diffusion law and
Mechanisms of Pitting Corrosion 271
Figure 16 Galvanostatic transient of Ni in saturated NiCl
2
solution with i = 1.5 A/cm
2
and the related voltage drop ΔE + ΔU
Ω
across the total salt layer and ohmic drop ΔU
Ω
across the porous film. (From Ref. 56.)
Copyright © 2002 Marcel Dekker, Inc.
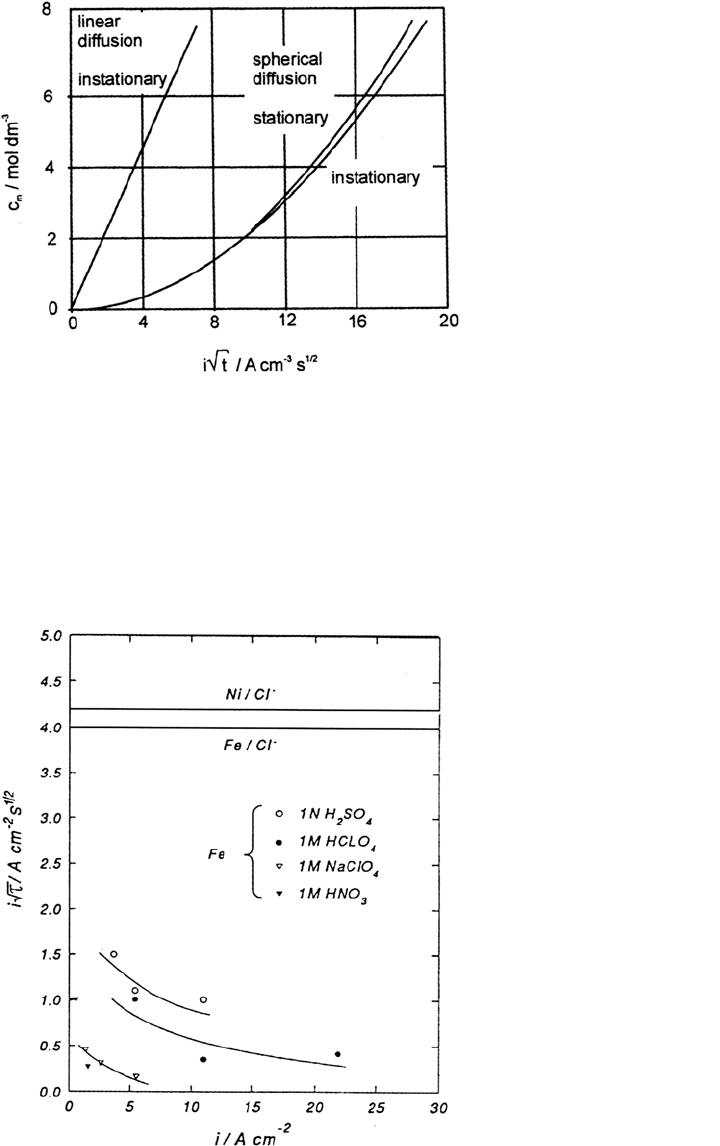
272 Strehblow
Figure 17 Concentration of NiCl
2
at a corroding Ni surface c
m
as a function of the product
i√t for linear and spherical transport (including nonstationary conditions) calculated with
Eqs. (17) and (20). (From Ref. 30.)
Figure 18 Plot of i – i√
τ
—
, τ = transition time, for galvanostatic transients of small Fe
electrodes in different solutions without and with 1 M Cl
–
depending on the applied current
density. (From Ref. 58.)
Copyright © 2002 Marcel Dekker, Inc.

Faraday’s law for the hemispherical electrode geometry with the same varibles as
used for Eq. (17):
Mechanisms of Pitting Corrosion 273
This equation contains a constant a that takes into account the change from the
convex to the concave pit geometry. For the simple analytical solution of the convex
hemispherical transport problem a = 1 will hold. For a concave hemispherical pit
a has been estimated to be 3 [6,30,31]. Computer-assisted numerical calculations
yield a result that is very close to this value for the pit bottom [62,63]. With the
simple stoichiometric relation between the local current density i
c,p
of a
hemispherical pit and its growth rate of the radius dr/dt and Faraday’s law [Eq.
(19)] one obtains Eq. (20), which is the equivalent relation for the transport from
a hemispherical pit to the linear transport from a small electrode dissolving totally
at its surface [Eq. (17)]. V
m
is the molar volume of the metal under study. Figure 17
compares the calculated results for NiCl
2
for both electrode geometries.
Precipitation of corrosion products is expected for the transition times t = τ when
saturation is achieved at the pit surface with c
m,s
= 4.8 M and 4.2 M for NiCl
2
and
FeCl
2
, respectively. This condition is achieved first at the pit bottom for a = 3. The
calculated data agree well with the experimentally observed precipitation within
pits [30]. For Ni
i√
τ
—
= 14.8 A cm
–2
s
1/2
is obtained. With the assumption of
nonstationary transport conditions one yields almost identical results (Fig. 17).
Some deviation is obtained only for values i√t
–
> 12 A cm
–2
s
1/2
[30].
The precipitation of a salt film is closely related to a change in the growth of
a corrosion pit. When the salt film is formed, the growth rate slows down and the
pit morphology changes from a polygonal to a hemispherical structure. This change
starts at the pit bottom, where it is expected first, and often is combined with an
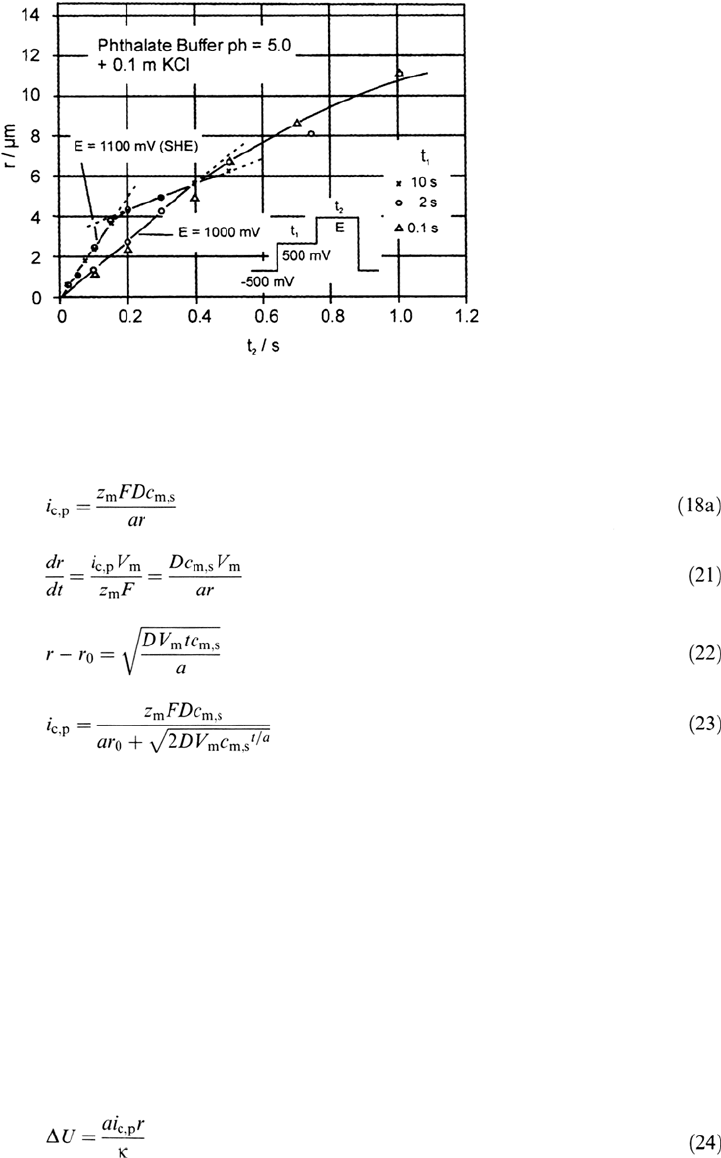
electropolishing effect [55]. Direct microscopic in situ and ex situ examinations of
growing pits for potentiostatic conditions show a decrease of the growth of the
radius when a salt film precipitates (Fig. 19) [58]. The time of change of the growth
rate coincides with the electropolishing effect at the pit bottom [55]. Heimgartner
and Böhni [64] discussed the growth of corrosion pits for longer times under
diffusion-limited conditions in more detail. If the local current density i
c,p
in a pit
is not constant and decreases inversely with the pit depth r according to Eq. (18a),
one deduces with Eq. (19) a reciprocal relation of its growth rate dr/dt with its
depth r [Eq. (21)]. Integration leads to a parabolic dependence of the pit radius on
time t [Eq. (22)] and combination with Eq. (18a) to Eq. (23) with an inverse change
of the local pit current density i
c,p
with √t
–
. This discussion assumes a saturated
solution with a change of the metal ion concentration Δc = c
m,s
, i.e., a salt film at
the actively corroding pit surface and a vanishing bulk concentration c
m,b
.
Therefore a later stage of pit growth is discussed and not the initial situation of free
metal dissolution as for the beginning of pit growth of Figure 15. Equations (22)
and (23) assume a minimum pit radius r
0
where a saturated solution at its surface
is achieved. Equation (23) is simply the combination of Eqs. (18a) and (22).
Copyright © 2002 Marcel Dekker, Inc.
Enhanced convection may lead to a situation where ohmic resistance instead of
diffusion is rate determining. Convection by an increasing flow rate of the electrolyte
or the evolution of gas bubbles in corrosion pits will increase the local dissolution
and finally cause ohmic current control. Like diffusion control, ohmic control will
lead to a parabolic dependence of the pit radius or pit depth on time.
Potential Drops
There exist detailed studies for the potential drop ΔU, the composition of the pit
electrolyte, and related changes of the pH. All these factors have been used to
explain the stability of a corroding pit. The potential drop within the electrolyte
for an open hemispherical pit can be estimated by the simple equation:
274 Strehblow
Figure 19 Deviation from linear pit growth with time when precipitation of a salt layer
occurs, Ni, E = 1.00 and 1.30 V phthalate buffer, pH 5.0 + 0.1 M KCl with inserted diagram
of applied potential pulses. (From Ref. 58.)
Copyright © 2002 Marcel Dekker, Inc.
with specific conductivity κ, local current density i
c,p
,
radius r, and geometric factor
a = 3 [6]. It depends on the specific situation whether ΔU is large enough to shift
the potential below the Flade potential, i.e., in the active range of the polarization
curve (Fig. 20). For 0.5 M H
2
SO
4
with κ = 0.22 cm
1
and i
c,h
= 0.3 A/cm
2
, which
is a reasonable value for moderately high pit current densities for Fe, one obtains
ΔU > 1 V for a pit radius r > 2.4 mm, but only ΔU = 0.41 mV for r = 1 μm. This
short calculation demonstrates that it is absolutely necessary to define well which state
of the growth of a pit is discussed. Potential drops may stabilize localized corrosion
for large pits but never for small ones, i.e., for initial stages. The conductivity of
the electrolyte is, of course, another factor that has to be taken into account. The
accumulation of ions within a corroding pit may increase
κ
and thereby reduce ΔU
compared with the value obtained with the bulk conductivity. Evidence for potential
control of pit growth is given for studies on Al by Hunkeler and Böhni [65]. They
found a linear dependence of the product i
c,p
r on the voltage drop ΔU.
Composition of Pit Electrolyte
The accumulation of electrochemically nonactive electrolyte components is closely
related to the potential drop ΔU within the electrolyte, similar to a Boltzmann
dependence of the concentration c
j
of these species on the electrical energy ΔU zF
[6,30]. Together with the electroneutrality equation, one obtains a dependence of
the concentration of the corroding metal ions c
m
at the metal surface on ΔU with
c
m
– c
mb
= c
m
for a vanishingly small bulk concentration c
mb
[6,30]. The
corresponding bulk values are c
j,b
and c
m,b
Mechanisms of Pitting Corrosion 275
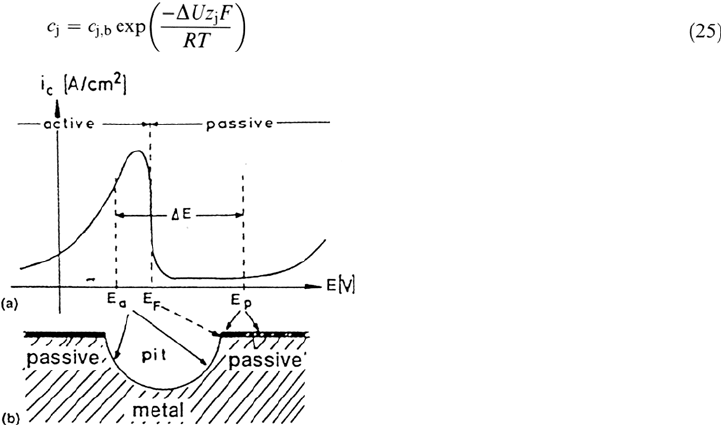
Figure 20 (a) Typical current density potential curve of a passive metal; (b) hemispherical
pit with potentials E
a
inside and E
p
outside the pit according to the rejected theory
frequently used for small pits. (From Ref. 31.)
Copyright © 2002 Marcel Dekker, Inc.
Equation (25) requires that only one electrochemical process occurs with no
subsequent reactions within the bulk electrolyte. The concentration of the metal
cations c
m
may also be calculated from Eq. (18) or (18a). For vanishingly small
bulk concentration of the cations, Δc equals the surface concentration c
m
. The
accumulation of cations is proportional to the local pit current density and the pit
radius [Eq. (18)]. One may therefore take i
cp
r as a measure of the cation
concentration at the pit surface, which depends on the age of the pit. Therefore this
scale is included in Figures 21 and 22.
276 Strehblow
The concentrations c
j
at the pit surface of species that are not involved in the
electrode process and the cation concentration have been calculated according to
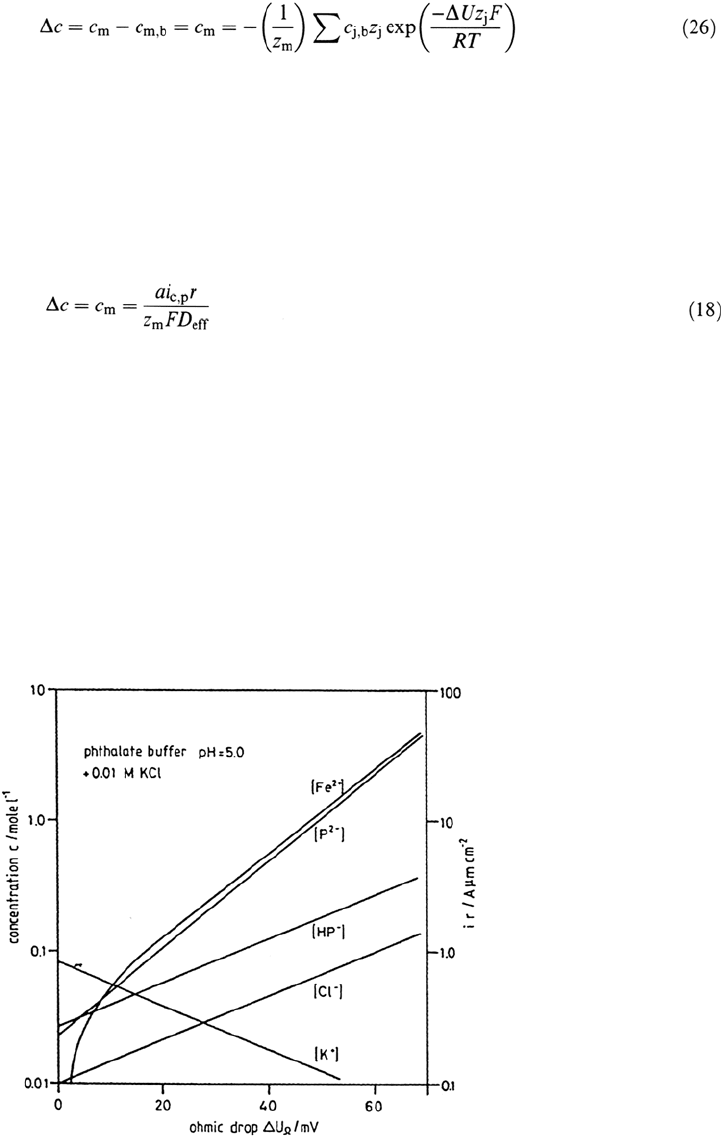
Eq. (25) for some bulk electrolytes frequently used for pitting studies (Fig. 21).
Metal ion concentrations c
m
go up to saturation of a few moles per liter when the
potential drop increases to several tens of millivolts, which is usually the case for
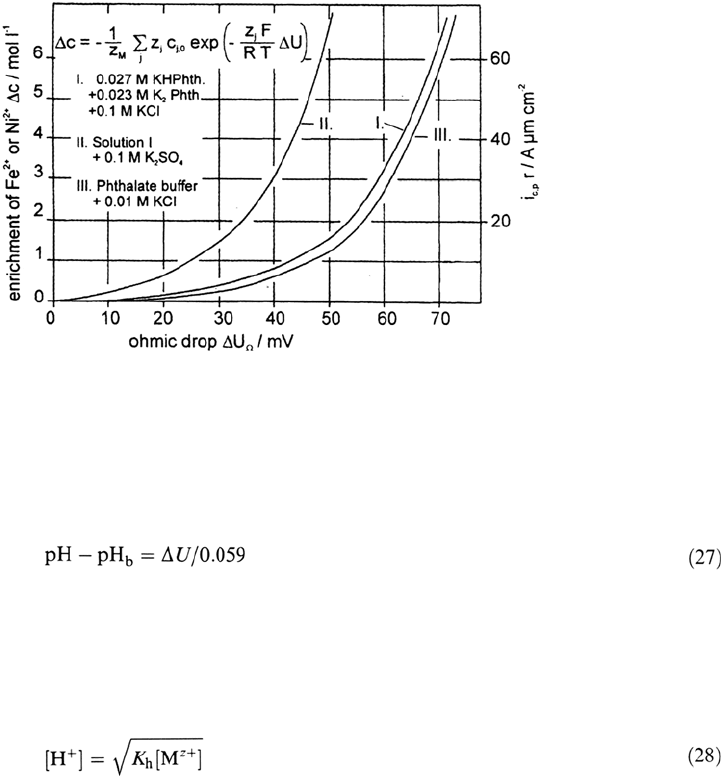
a pit radius of several micrometers. Equation (26) was applied for the calculation
of the concentrations c
m
at the pit surface as given in Figure 22.
pH shifts have often served to explain the stability of a corroding pit. Equation
(25) permits the calculation for buffered solutions when the pH is not affected by
hydrolysis of the corrosion products [30]. Buffer ions change in concentration
in agreement with the pH shift as given for the phthalate buffer in Figure 21. An
Figure 21 Ionic concentration at the metal surface depending on the ohmic drop ΔU
Ω
within the electrolyte according to Eqs. (25) and (26). (From Ref. 30.)
Copyright © 2002 Marcel Dekker, Inc.
accumulation of dissolving metal ions leads to accumulation of the (buffer) anions
and to depletion of the other cations and consequently to a positive pH shift
according to Eq. (27).
Mechanisms of Pitting Corrosion 277
Figure 22 Concentration of metal ions at the dissolving metal surface depending on the
ohmic drop ΔU
Ω
and the parameter i
c,p
r according to Eq. (26). (From Ref. 30.)
In nonbuffered solutions the usual hydrolysis equilibrium of the metal ions leads
to acidification according to
Me
z+
+ H
2
O ↔ MeOH
(z–1)+
+ H
+
With [MeOH
(z–1)+
] = [H
+
] for nonbuffered electrolytes one obtains:
With hydrolysis constants of K
h
= 10
–10
and 10
–7
one obtains for the saturation
concentrations 4.8 and 4.2 M, pH values of 4.3 and 2.4 for saturated NiCl
2
and FeCl
2
solutions, respectively. These calculations show that the precipitation of hydroxide
within a corrosion pit may be prevented for these metals by acidification. However,
as discussed before, passivation should nevertheless be possible for many technically
important metals. For Fe, Ni, and other metals, especially in acidic media, passivity
cannot be explained on the basis of thermodynamic equilibria and Pourbaix
diagrams. In these cases passivity is a kinetic phenomenon. Otherwise these metals
should not show passivity in a strongly acidic environment. Thus, different
explanations are required for the stability of a corrosion pit.
The pH shift of the passivation potential E
pa
may not give an explanation either.
Its change usually by –0.059 V/pH is much too small to cause a shift above the
potentiostatically applied value E well within the passive range, so that the metal
surface might reach the active range of the polarization curve. In 0.5 M H
2
SO
4
the
passivation potential for Fe or Ni is E
pa
= 0.58 or 0.35 V, respectively, with a shift of
–0.059 V per pH unit. If the potential is set in the passive range up to 1.5 V one has
Copyright © 2002 Marcel Dekker, Inc.

to explain a shift of the passivation potential by 0.9 to 1.15 V, which is absolutely out
of range of any reasonable explanation. If, however, the potential E is set to values
close to E
pa
, as often realized in technical corrosion situations with open-circuit con-
ditions, acidification may cause its shift above E with E
pa
> E, which especially in
weakly acidic and alkaline unbuffered solutions may fulfill these requirements. In
addition, the much more negative passivation potential of Fe for neutral and alkaline
solutions, such as E
pa
= –0.1-0.059 pH for Fe, will not be effective as a consequence
of acidification by hydrolysis. The passivation potential becomes more negative
because of the insolubility of lower valent Fe oxides such as Fe
3
O
4
or Fe(OH)
2
in
neutral and alkaline solutions. However, these oxides dissolve very quickly in acidic
electrolytes, so they do not provide passivity for these conditions. In conclusion, one
may say that local negative pH shifts due to hydrolysis of corrosion products may
support and stabilize pitting at negative electrode potentials for neutral and weakly
acidic and alkaline bulk electrolytes. This will hold especially for metals that cannot
be passivated at low pH, e.g., Cu and Al, because of their fast dissolution or high
solubility in this environment. However, this interpretation does not hold for Fe, Ni,
and steels in strongly acidic electrolytes at sufficiently positive electrode potentials.
Thus, pitting needs another, more general explanation. Furthermore, anions of other
strong acids such as perchlorates and sulfates should act as aggressive anions as well
if hydrolysis and pH shifts are the essential causes of localized corrosion. However,
ClO
4
–
may even act as an inhibitor if the potenital is not too positive and most
metals may be passivated in sulfuric and perchloric acid, which demonstrates that
the specific chemical properties of the aggressive anions have to be taken into
account in the explanation of pitting corrosion.
An attempt has been made to explain the requirement for a minimum
concentration of aggressive anions to maintain stable pit growth [30,31]. With the
assumption that a salt film of thickness δ has to be maintained at the growing pit
surface, one obtains:
278 Strehblow
With this assumption the experimentally found value of c
min
= 3 × 10
–4
M has been
confirmed with a local current density i
c,p
= 1 A/cm
2
, a salt layer thickness δ = 5 nm,
geometric factor a = 3, molar volume V
m
= 3.55 cm
3
/val for the equivalent volume
for Fe metal and V
s
= 21.25 cm
3
/val for FeCl
2
, and diffusion constant
D =10
–5
cm
2
/s for Cl
–
ions.
The accumulation of aggressive anions has been found at the surface even of
small polygnal pits of some few μm diameter when their change to a hemisphere by
the precipitation of a salt film was not already achieved. A special preparation
technique of pulling the electrode with actively corroding pits into a benzene layer
above the electrolyte preserved the special situation at the corroding pit surface even
after rinsing with acetone and dry storage in air [66]. These pits remained active and
continued their growth immediately when reintroduced into the electrolyte at the
same potential; however, they became inactive on rinsing with water or stepping
the potential below the critical value. With electron microprobe analysis, chloride
could be found that corresponded to layer of ~ 5 nm FeCl
2
when the pit remained
active but was lost completely on rinsing with water or repassivation [66]. These
studies show clearly that the formation of a thin layer of aggressive anions even in
pits of some few μm is responsible for stable pit growth. The later precipitation
Copyright © 2002 Marcel Dekker, Inc.