Gupta D. (Ed.). Diffusion Processes in Advanced Technological Materials
Подождите немного. Документ загружается.


higher temperature, about 1200 K. This feature was explained
[52]
by the
contribution of short-circuit diffusion, since polycrystalline material was
used. Frank et al.
[53]
thoroughly measured Ni self-diffusion, combining the
tracer technique at higher temperatures and the SIMS analysis at lower
ones in poly- and single-crystalline Ni
3
Al, respectively. No curvature of
the Arrhenius plot was established [see Fig. 4.8(a)], confirming the expla-
nation given by Shi et al.
[52]
These data indicate that only one diffusion
mechanism operates in the temperature interval under consideration. This
mechanism is commonly identified with the Ni-sublattice diffusion mech-
anism.
[52, 53]
According to this mechanism, a Ni vacancy performs random
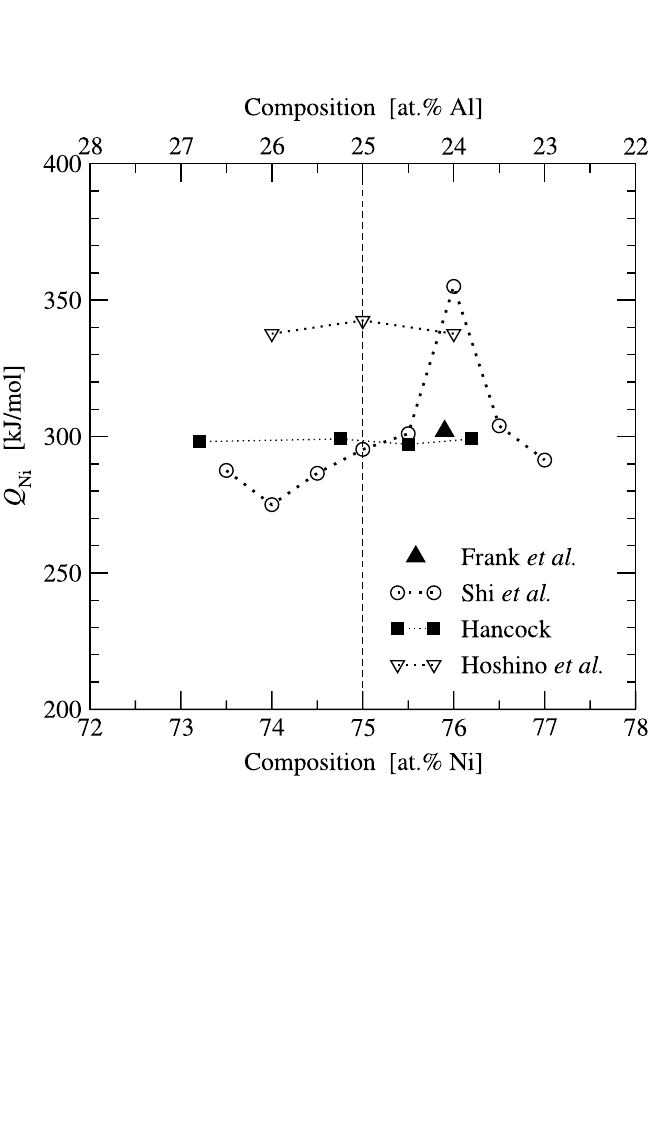
jumps in the Ni sublattice with the coordination number 8 (see Fig. 4.5).
Some controversy still exists about the compositional dependence of
Ni self-diffusion in Ni
3
Al (Fig. 4.9). Hoshino et al.
[51]
have found a shal-
low minimum of D
*
Ni
at the stoichiometric composition below T 1100 K.
Shi et al.
[52]
also observed such a minimum in D
*
Ni
, but around 76 at.% Ni
and, again, at the lowest temperature of their investigations, about 1200 K.
At higher temperatures, the variation of D
*
Ni
with composition in all inves-
tigations was within experimental uncertainities. These fine features
resulted in a maximum activation enthalpy Q
Ni
at about 76 at.% Ni
[52]
(Fig. 4.9). Since Ni grain boundary diffusion in Ni
3
Al shows a deep min-
imum at the stoichiometric composition, Frank and Herzig
[55]
suggested
that short-circuit diffusion via grain boundaries affected the low-temperature
data of Hoshino et al.
[51]
and Shi et al.,
[52]
and produced a slight minimum
in the bulk diffusion data. Comparing results of different investigations,
Fig. 4.9 suggests that the change of the activation enthalpy Q
Ni
with com-
position in Ni
3
Al is within experimental uncertainties. In contrast, a min-
imum in D
*
Ni
was found in a recent Monte Carlo study of Ni diffusion in
Ni
3
Al at the stoichiometric composition
[56]
due to the existence of Al and
Ni antistructure atoms in Ni- and Al-rich alloys, which enhance Ni self-
diffusion by lowering the energy barriers for the Ni vacancy jumps. In that
approach, pair interaction potentials were used and no lattice relaxation
was taken into account.
For additional insight into this problem, we simulated self-diffusion
in Ni
3
Al using the Voter and Chen EAM potentials.
[24]
The calculations
show that there is a negligible effect of adjacent Ni and Al antistructure
atoms on the energy barriers for Ni vacancies performing jumps via the
Ni sublattice. (The energy barriers change by about 10 kJ/mol, depend-
ing on the particular configuration.) Thus, the vacancy jump rates are
nearly the same, regardless of the composition of Ni
3
Al. Calculations of
the Ni vacancy concentration [Fig. 4.3(c)] and the effective formation
energies also resulted in very similar values for Al-rich, stoichiometric,
and Ni-rich compounds. Simultaneous Monte Carlo simulations of the
correlation effects demonstrated that the correlation factor for the Ni
192 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
Chapter-04.qxd 11/29/04 6:36 PM Page 192
DIFFUSION IN INTERMETALLIC COMPOUNDS, HERZIG, DIVINSKI 193
sublattice diffusion mechanism across the stoichiometry changes negligi-
bly and equals about 0.689. These calculations support the conclusion that
Ni self-diffusion reveals only a marginal compositional dependence in
Ni
3
Al.
[53]
From the experimental point of view, Ni diffusion in the Ni
3
Al
phase can be presented by a unique Arrhenius line with the parameters
given in Table 4.1.
Al Diffusion.
26
Al radiotracer diffusion in Ni
3
Al was measured
[41]
at
temperatures between 1273 and 1473 K. Unfortunately, the absolute val-
ues of Al diffusion are considered to be unconvincing because there is a
Figure 4.9 Activation enthalpy Q
Ni
of Ni self-diffusion in Ni
3
Al as a function of com-
position (open circles,
[52]
triangle up,
[53]
triangles down,
[51]
and squares
[54]
).
Chapter-04.qxd 11/29/04 6:36 PM Page 193

194 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
substantial deviation of the simultaneously measured Ni self-diffusion
results from the data of Frank et al.
[53]
Incomplete consideration of short-
circuit diffusion in polycrystalline samples may be one possible reason
for such deviations. However, the ratio of Al and Ni diffusivities meas-
ured by Larikov et al.
[41]
is likely to be more reliable, and we can antici-
pate that Al and Ni diffuse with similar rates in Ni
3
Al in this temperature
interval.
The interdiffusion coefficient D
∼
accross the phase field of Ni
3
Al was
measured by two groups.
[57, 58]
The temperature dependence of D
∼
for stoi-
chiometric Ni
3
Al is presented in Fig. 4.8(a). Intrinsic diffusivities of Al
and Ni were determined between 1425 and 1523 K.
[59]
Analyzing the rel-
ative positions of the Kirkendall and Matano planes, Fujiwara and Horita
concluded that the ratio of Al and Ni tracer diffusivities is very close to
unity in this temperature interval,
[59]
supporting the finding of Larikov
et al.
[41]
In Fig 4.8(a), the estimated Al diffusion coefficients are compared
with the Ni self-diffusivity. On the other hand, the Al diffusivity was esti-
mated by Ikeda et al.
[57]
using the Darken-Manning equation [Eq. (3); see
Fig. 4.8(a)]. Their smaller values of D
*
Al
with respect to Fujiwara and
Horita’s results
[59]
may stem from some uncertainty involved in the calcu-
lations of the product Φ S.
Similar to Ni tracer diffusion, the interdiffusion coefficient in Ni
3
Al
reveals only a marginal compositional dependence, which remains within
Table 4.1. Arrhenius Parameters of Self-Diffusion and Solute Diffusion in
Ni
3
Al
Composition D
0
Q
Tracer (at.% Ni) (m
2
s
1
) (kJ/mol) Ref.
Ni 75 1.5 10
2
347 51
Ni 74.7 1.0 10
4
303 54
Ni 75.9 3.1 10
4
302 53
Al
*
75 8 10
3
340
Ga 75.9 7.8 10
2
363 63
Ge 75.9 1.0 10
1
369 63
Ge 75 3.6 10
1
379 61
Nb 75.9 2.6 10
1
476 63
Ti 75.9 8.6 10
1
425 63
Mo 75.1 2.3 10
2
493 61
Fe 75.1 1.3 10
2
335 64
*
Estimation from the Al intrinsic diffusion data of Fujiwara and Horita
[59]
Chapter-04.qxd 11/29/04 6:36 PM Page 194

DIFFUSION IN INTERMETALLIC COMPOUNDS, HERZIG, DIVINSKI 195
the experimental uncertainties.
[59]
From this we conclude that the Al dif-
fusivity does not markedly depend on composition in Ni
3
Al.
4.4.1.2 Solute Diffusion
Since Ge and Ga occupy the Al sublattice in Ni
3
Al,
[60]
these tracer
atoms can be used as the substitute for the Al diffusion.
[38, 61]
The available
data are presented in Fig. 4.8(b). Their diffusivities follow very similar
and straight Arrhenius dependencies in Ni
3
Al, with an activation enthalpy
of about 60 kJ/mol larger than that of Ni self-diffusion. It is important that
Ga and Ge diffusion in Ni
3
Al is very close to the Al diffusivity estimated
from the interdiffusion data
[59]
[Fig. 4.8(b)]. We conclude that these solute
atoms are indeed useful to simulate Al diffusion in Ni
3
Al.
Figure 4.8(b) suggests D
*
Ga
D
*
Ni
at lower temperatures. This relation
is reversed, however, at higher temperatures, T 1350 K, where D
*
Ga
D
*
Ni
. This trend can also be anticipated for Al diffusion [see Fig. 4.8(b)].
Such behavior cannot be understood in terms of the Ni sublattice diffusion
mechanism of the minority component, as suggested by Numakura
et al.
[35]
Indeed, the effectiveness of diffusion by this mechanism strongly
depends on the probability of finding an Al (or Al-substitute) atom on the
wrong Ni sublattice. Consequently, Al should diffuse slower than Ni, in
agreement with the so-called Cu
3
Au rule.
[62]
However, it was shown by
Divinski et al.
[38]
that the ASB diffusion mechanism provides an addi-
tional and strong contribution for Al atoms comparable to the Ni sublat-
tice diffusion mechanism and that D
*
Al
can approach the D
*
Ni
values at high
temperatures, in accordance with the experimental findings of Fujiwara
and Horita
[59]
[Fig. 4.8(b)].
Tracer diffusion of Ti and Nb was investigated
[63]
[Fig. 4.8(b)]. In con-
trast to Ge and Ga diffusion, Ti and Nb reveal a distinctly smaller diffu-
sivity with a larger activation enthalpy. From the direction and extent of the
homogeneity range of the L1
2
phase in a ternary region, the site preference
for Ti and Nb in Ni
3
Al was estimated.
[60]
Because Ti and Nb atoms mainly
dissolve on Al positions in the L1
2
lattice, a diffusion process similar to that
of Al is to be expected. The difference in the activation enthalpies of self-
diffusion and diffusion of such solutes is related to the formation energy of
the solute atom at an antistructural position in the Ni sublattice.
[53]
In Fig. 4.8(b), the tracer diffusivities of Mo
[61]
and Fe
[64]
in Ni
3
Al are
also plotted, as functions of the inverse temperature. The solute diffusion
behavior in Ni
3
Al can be well systemized in terms of an interplay of intra-
sublattice (the Ni sublattice mechanism) and inter-sublattice (the ASB
mechanism) jumps.
[61]
The Arrhenius parameters of self- and solute diffu-
sion in Ni
3
Al are summarized in Table 4.1.
Chapter-04.qxd 11/29/04 6:36 PM Page 195

196 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
4.4.2 Ti
3
Al
4.4.2.1 Self-Diffusion
Ti Diffusion. Self-diffusion and solute diffusion in Ti
3
Al was com-
prehensively analyzed in a recent review.
[8]
In an investigation of Ti self-
diffusion in the phase a
2
-Ti
3
Al,
[65]
several compositions were studied: 75,
72, 68, and 65 at.% Ti. Within experimental error, the diffusivities were
very similar, with a slight increase of D
*
Ti
with increasing Al content at the
lowest temperature investigated. The change of Al content produced only
a marginal effect in the activation enthalpy of self-diffusion, Q
Ti
, although
the tendency of an increase of Q
Ti
with increasing Al content was indi-
cated.
[65]
As a result, one master Arrhenius dependence for Ti self-diffusion
in a
2
-Ti
3
Al was suggested [Fig. 4.10(a)] with the Arrhenius parameters
given in Table 4.2.
Ti self-diffusion in Ti
3
Al can be interpreted within the framework of
the Ti-sublattice diffusion mechanism.
[65]
The experimental data suggest
that the compositional Ti or Al antistructure atoms change the energy bar-
riers for vacancy jumps on the Ti sublattice only slightly. Since the Ti
vacancy formation enthalpies are also very similar for the stoichiometric
and Al-rich alloys,
[8, 19]
the weak compositional dependence of D
*
Ti
in Ti
3
Al
is not unexpected.
Al Diffusion. Interdiffusion in the a
2
-Ti
3
Al-phase field was stud-
ied,
[65, 66]
with good agreement of the deduced diffusion data, which
practically overlap in Fig. 4.10(a). Applying the Darken-Manning analy-
sis with S 1 [Eq. (3)], Fig. 4.10(a) presents the calculated Al diffusion
coefficient D
*
Al
as a function of temperature. The thermodynamic factors
required for such calculations were calculated by the CALPHAD
Table 4.2. Arrhenius Parameters of Self-Diffusion and Solute Diffusion in
Ti
3
Al
D
0
Q
Tracer (m
2
s
1
) (kJ/mol) Ref.
Ti 2.24 10
5
288 65
Al
*
2.3 10
1
394 65
Ga 6.3 10
5
315 68
Fe 2.0 10
3
277 70
Ni 1.8 10
5
195 70
Nb 3.2 10
4
339 70
*
Calculation from the Darken-Manning equation
Chapter-04.qxd 11/29/04 6:36 PM Page 196
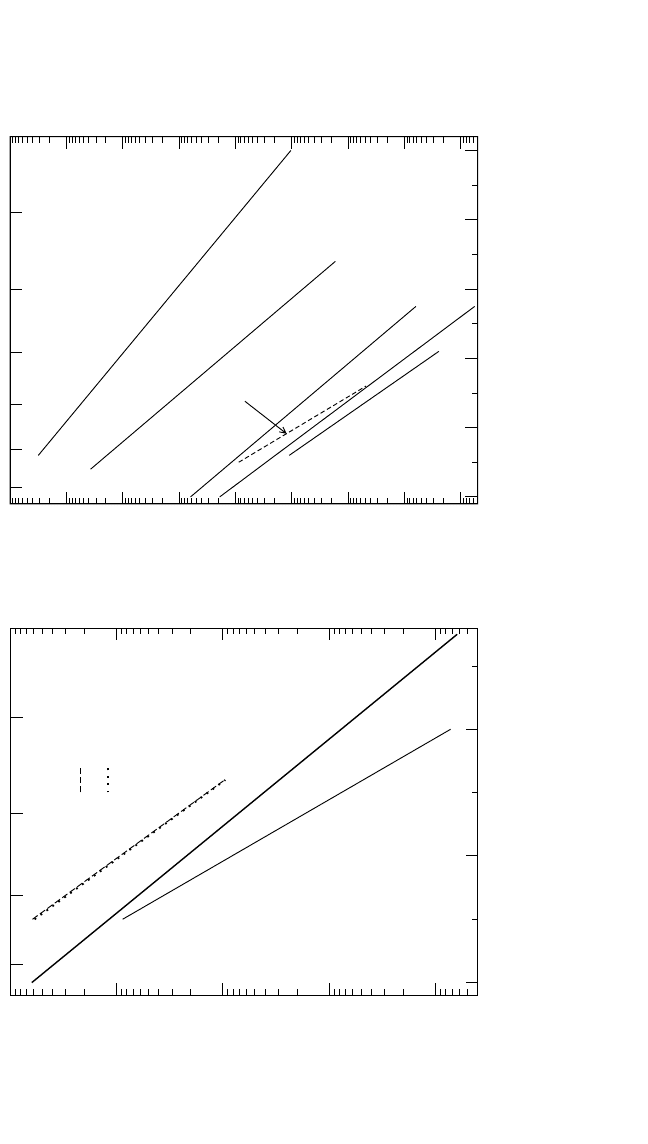
197
7
89
T
-1
[10
-4
K
-1
]
10
-19
10
-18
10
-17
10
-16
10
-15
D
*
, D [m
2
s
-1
]
Rüsing et al.
Sprengel et al.
1400 1300 1200 1100
T [K]
~
Al
Ti
interdiffusion:
Figure 4.10 The Arrhenius plots of self-diffusion and solute diffusion in Ti
3
Al. In (a), the interdiffusion data D
∼
[66]
and
the estimated Al diffusivity
[65]
are plotted in comparison to Ti diffusion.
[65]
In (b), diffusion data for Ti,
[65]
Al,
[66]
Ga,
[68]
and
Nb, Fe, and Ni
[70]
diffusion are compared.
7
891011 12
T
-1
[10
-4
K
-1
]
10
-20
10
-19
10
-18
10
-17
10
-16
10
-15
10
-14
10
-13
10
-12
D
*
[m
2
s
-1
]
1400 1300 1200 1100 1000
900
T [K]
Nb
Fe
Ni
Ga
Ti
Al
(b) Solute Diffusion in Ti
3
Al
(a) Self-Diffusion in Ti
3
Al
Chapter-04.qxd 11/29/04 6:36 PM Page 197

198 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
method.
[67]
The weak compositional dependence of D
*
Al
in Ti
3
Al is within
experimental error, but a slight increase with increasing Al content is
indicated.
[8]
The activation enthalpy Q
Al
of Al diffusion ≈400 kJ/mol
(Table 4.2) turned out to be fairly large. The analysis of the experimen-
tal data suggests that the large value of Q
Al
may be related to uncertain-
ties in the D
*
Al
determination via the Darken relation, especially at low
temperatures.
[68]
4.4.2.2 Solute Diffusion
Since Ga occupies the Al sublattice in Ti
3
Al,
[69]
the study of Ga diffu-
sion is meaningful to obtain additional information on diffusion of an Al-
substituting solute in Ti
3
Al.
[68]
The measured Ga diffusion coefficients are
presented in Fig. 4.10(b) as a function of temperature. The figure indicates
that the absolute values of D
*
Ga
are similar to those of D
*
Al
, but the activa-
tion enthalpy of Al diffusion is distinctly larger (see Table 4.2).
Ga diffusion in Ti
3
Al was interpreted in terms of the Ti-sublattice dif-
fusion mechanism of the minority component.
[68]
Thus, the excess of Q
Ga
with respect to Q
Ti
is related to the formation enthalpy of Ga atoms in anti-
structure positions on the Ti sublattice.
[68]
Due to the predominant vacancy
concentration in the Ti sublattice [Fig. 4.3(a)], Al is also expected to dif-
fuse predominantly as anti-structure atoms via the Ti sublattice. Thus we
regard the measured Q
Ga
value as realistic to represent Q
Al
in Ti
3
Al.
Information is limited on diffusion of other solutes in a
2
-Ti
3
Al. Diffusion
of Nb in Ti
3
Al, studied
[70]
within the composition range from 27 to 33 at.%Al,
was found to be practically independent of composition. The corresponding
temperature dependence is presented in Fig. 4.10(b). Nb atoms occupy Ti sites
in Ti
3
Al.
[69]
Therefore, their diffusion is dominated by exchanges with vacan-
cies on the Ti sublattice (Ti-sublattice diffusion mechanism).
In view of the extremely fast diffusivity in a-Ti, diffusion of Fe and
Ni was investigated in Ti
3
Al.
[70]
Only a negligible compositional depend-
ence was observed, similar to the diffusion of Ga and Nb. The tempera-
ture dependencies obtained are presented in Fig. 4.10(b). This diagram
shows that Fe and Ni are also fast diffusers in Ti
3
Al, since their diffusivi-
ties exceed that of Ti self-diffusion by several orders of magnitude.
Fast diffusion of Fe and Ni in Ti
3
Al was explained
[8, 70]
by the disso-
ciative diffusion mechanism; that is, the diffusing atoms occupy both
interstitial and substitutional sites. In such a case, the effective diffusion
coefficient D is given by:
D D
i
D
s
D
i
. (5)
c
i
c
s
c
s
c
i
c
s
c
i
c
i
c
s
Chapter-04.qxd 11/29/04 6:36 PM Page 198

DIFFUSION IN INTERMETALLIC COMPOUNDS, HERZIG, DIVINSKI 199
Here D
i
(D
s
) is the interstitial (substitutional) diffusion coefficient, and
c
i
(c
s
) is the interstitial (substitutional) solubility of the solute atoms.
Equation (5) was simplified under the plausible assumptions that c
i
c
s
and c
i
D
i
c
s
D
s
. Since c
i
and c
s
are temperature-dependent, the activa-
tion enthalpy of the dissociative diffusion can be remarkably larger than
that of pure interstitial diffusion. This conclusion agrees with the obser-
vation that the activation enthalpies of Fe and Ni diffusion (277 and
195 kJ/mol, respectively), in comparison with the Ti self-diffusion activa-
tion enthalpy (288 kJ/mol), are not as small as expected for the pure inter-
stitial diffusion mechanism.
4.4.3 TiAl
4.4.3.1 Self-Diffusion
Ti Diffusion. The self-diffusion behavior in Ni
3
Al and Ti
3
Al follows
almost ideal linear Arrhenius dependencies (see Figs. 4.8 and 4.10,
respectively), a behavior that is typical for the majority of intermetallic
phases. From a simplest point of view, such a behavior is somewhat unex-
pected in view of the variety of point defects and the number of possible
diffusion mechanisms in ordered intermetallics. However, the experimen-
tal data suggest that in many typical intermetallic phases, there is a given
diffusion mechanism that produces the main contribution to self-diffusion
and operates dominantly over a wide temperature interval. Astonishingly,
Ti self-diffusion in g-TiAl clearly revealed a non-Arrhenius character
[7]
[see Fig. 4.11(a)]. The Ti diffusivity follows an Arrhenius line up to about
T 1470 K, with a subsequent upward deviation to larger D
*
Ti
values at
higher temperatures. This effect was found to be almost independent of
the composition in TiAl.
[7]
Table 4.3 presents the activation enthalpy Q
Ti
of Ti-self diffusion in TiAl calculated by the Arrhenius fitting within the
temperature interval 1150 to 1470 K.
The pronounced curvature of Ti self-diffusion at T 1470 K is
explained by the additional effect of a second diffusion mechanism that
operates parallel to the Ti-sublattice mechanism.
[7]
In view of the high
concentration of Ti antistructure atoms, even in Al-rich compositions [see
Fig. 4.3(b)], the antistructure bridge mechanism [Fig. 4.7(b)] was pro-
posed to produce this additional contribution. The most important feature
of the ASB mechanism, when operating in the L1
0
structure of TiAl, is that
it reveals a nonpercolative nature and thus contributes at any concentra-
tion of the Ti antistructure atoms. This effect results from the interde-
pendence of the ASB mechanism with the simultaneous contribution of
the Ti-sublattice diffusion mechanism. Thus, in TiAl, the situation is very
different from that in the B2 compounds, where the percolation threshold
Chapter-04.qxd 11/29/04 6:36 PM Page 199
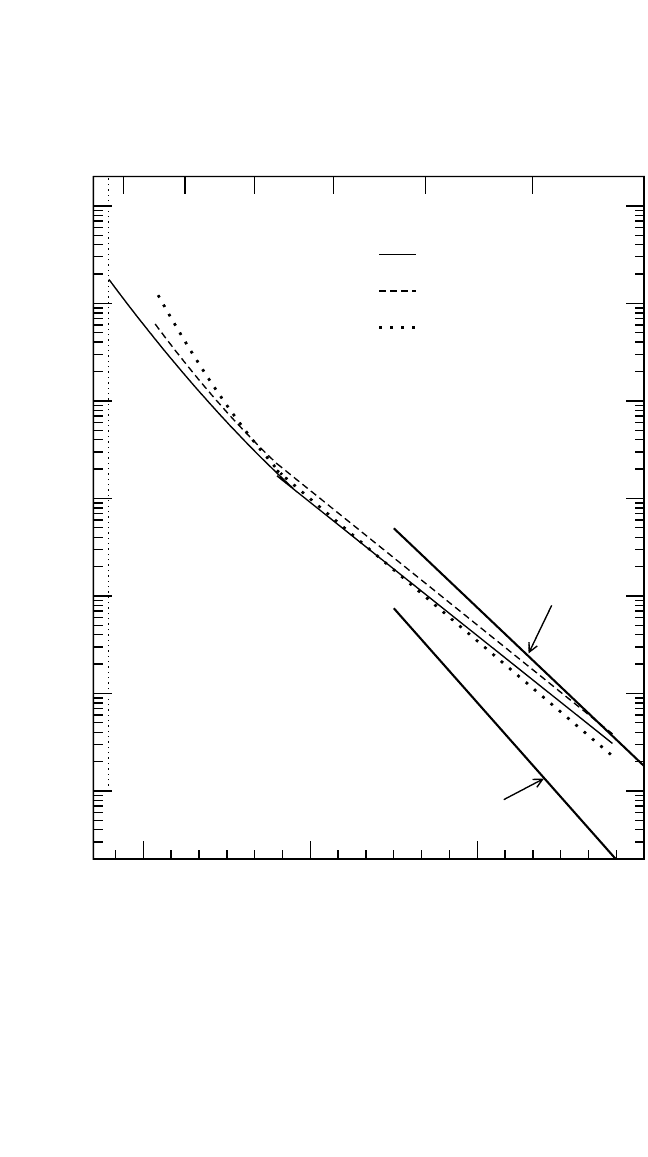
200 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
Figure 4.11 The Arrhenius presentation of Ti and Al component and solute diffu-
sion in TiAl. In (a), Ti self-diffusion in polycrystalline TiAl alloys of the indicated
compositions
[7]
is plotted in comparison to Ti diffusion along (D
Ti
) and perpendi-
cular (D
⊥
Ti
) to the Ti layers in the L1
0
structure of TiAl.
[71]
In (b), the interdiffusion
data,
[72]
estimated Al
[7]
and Ga
[68]
diffusivities are shown.In (c), Ti;
[7]
Nb, Zr, Fe, and
Ni;
[39]
and Co and Cr
[74]
diffusion is presented.
6
7
89
T
-1
[10
-4
K
-1
]
10
-18
10
-17
10
-16
10
-15
10
-14
10
-13
10
-12
D
*
[m
2
s
-1
]
Ti in Ti
44
Al
56
Ti in Ti
46
Al
54
Ti in Ti
47
Al
53
1500 1400 1300 120016001700
T [K]
D
⊥
Ti
D
||
Ti
(a) Ti Component in TiAl
Chapter-04.qxd 11/29/04 6:36 PM Page 200
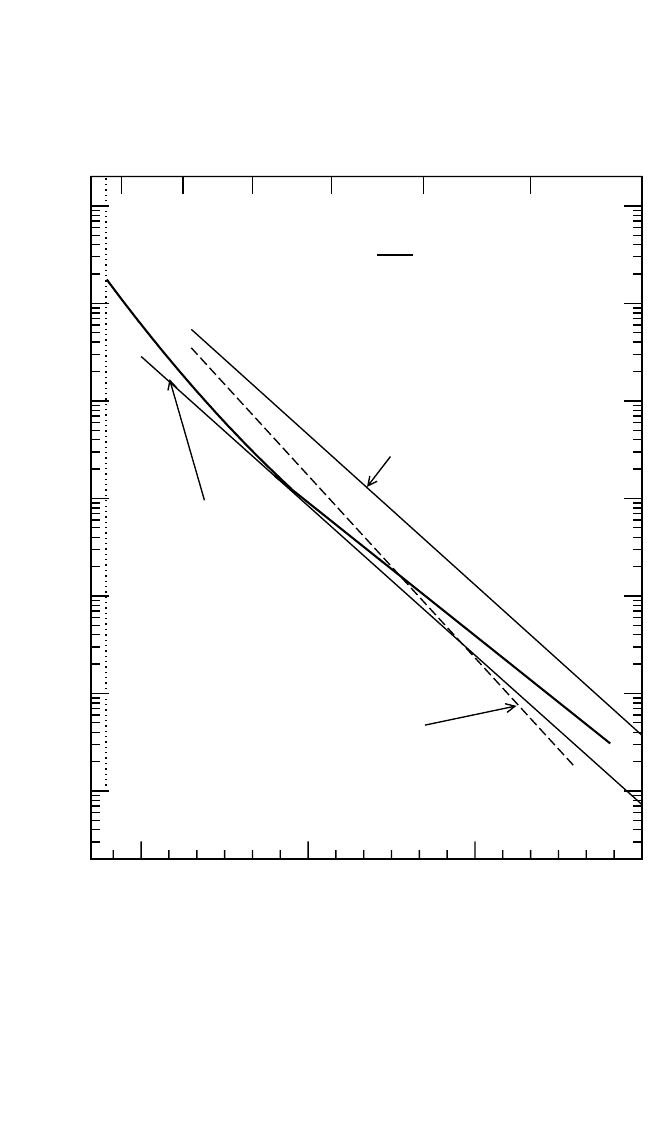
DIFFUSION IN INTERMETALLIC COMPOUNDS, HERZIG, DIVINSKI 201
6
7
89
T
-1
[10
-4
K
-1
]
10
-18
10
-17
10
-16
10
-15
10
-14
10
-13
10
-12
D
*
, D [m
2
s
-1
]
Ti in Ti
44
Al
56
1500 1400 1300 120016001700
T [K]
~
D (Sprengel et al.)
Al
~
Ga
(b) Al Component in TiAl
Figure 4.11 (Continued)
Chapter-04.qxd 11/29/04 6:36 PM Page 201