Jones M., Fleming S.A. Organic Chemistry
Подождите немного. Документ загружается.


14.13 Special Topic: Stable Carbocations in “Superacid” 679
the presence of an acid chloride (here, SO
2
Cl
2
), in order to produce a good
leaving group. This reaction converts the oxidized pyridine into the substituted
nonoxidized pyridine.
Nature uses a similar reaction in biological oxidation, the transfer of hydride to
the para position of the pyridinium ion, NAD
, the oxidized form of nicotinamide
adenine dinucleotide. We will see more of this reaction in Chapter 16 (Fig. 14.108).
+
..
O
..
..
C
hydride
transfer
NH
2
..
..
O
..
..
C
N
RR
H
NAD
+
N
–
..
O
..
C
NH
2
..
H H
FIGURE 14.108 Reduction of NAD
through hydride transfer.
Summary
There are similarities between nucleophilic aromatic substitution (S
N
Ar) and its
more usual counterpart, electrophilic aromatic substitution.Each involves the for-
mation of a resonance-stabilized intermediate,and each involves a temporary loss
of aromaticity that is regained in the final step of the reaction. But the similar-
ities are only so deep.The electrophilic reaction involves cationic intermediates;
the nucleophilic involves anionic intermediates. Use the differing effects of a
nitro group, strongly deactivating in the electrophilic substitution and strongly
activating in the nucleophilic substitution, to keep the two mechanisms distinct
in your mind.
SbF
5
–
SbF
6
nonnucleophilic
solvent, low
temperature
F
R
+
R
+
FIGURE 14.109 Cation formation
through reaction of an alkyl fluoride
with SbF
5
.
14.13 Special Topic: Stable Carbocations
in “Superacid”
The Friedel–Crafts reactions succeed because relatively stable carbocations can be
formed by the reaction of aluminum chloride (an exceptionally strong Lewis acid with
which an alkyl halide will react) in solvents that are nonnucleophilic. The strongest
nucleophile available in the reaction vessel is benzene, which attacks the cation to
start the Friedel–Crafts reaction. If stable carbocations cannot be formed, as with
methyl chloride or ethyl chloride, the AlCl
3
complexes can also alkylate benzenes.
How would we modify the reaction conditions so as to maximize the possibil-
ity of generating the carbocation and minimize the chances for its further reaction?
Antimony pentafluoride (SbF
5
) is an even stronger Lewis acid than AlCl
3
toward
halogens, particularly fluorine. When SbF
5
removes a fluoride ion from an alkyl
compound the anion antimony hexafluoride (SbF
6
) is produced (Fig.14.109).This
anion is nonnucleophilic, and fluoride ion is a poor nucleophile as well.

680 CHAPTER 14 Substitution Reactions of Aromatic Compounds
HC F
C
F
H
3
C
H
3
C
H
3
C
HC
+
H
3
C
H
3
C
CH
3
CH
3
C
+
H
3
C
CH
3
CH
3
SbF
5
–
SbF
6
–
SbF
6
SO
2
ClF, –60 ⬚C
SbF
5
SO
2
ClF, –60 ⬚C
FIGURE 14.110 Stable secondary and tertiary carbocations can be formed in superacid.
Many carbocations,once thought to be impossible species even as reaction inter-
mediates, can be generated and studied under superacid conditions.This work was
pioneered by George A. Olah (b. 1927) and earned Olah the Nobel Prize in
Chemistry in 1994.In future chapters, we will see a number of examples of the util-
ity of looking at cations in superacid.
K
+ –
NH
2
NH
3
, –33 ⬚C
Cl
NH
2
Aniline
(50%)
+ KCl
Chlorobenzene
FIGURE 14.111 Aniline can be
formed from chlorobenzene by
reaction with very strong base,
potassium amide in ammonia.
PROBLEM 14.39 Explain why SbF
6
is nonnucleophilic.
Treatment of alkyl fluorides with SbF
5
at very low temperatures in highly polar, non-
nucleophilic solvents such as SO
2
,SO
2
ClF,or FSO
3
H (so-called superacid conditions)
can generate many stable carbocations. This process is Friedel–Crafts chemistry
carried to extremes. Methyl and primary carbocations cannot be generated this way,
but tertiary and secondary cations can be as long as they are not prone to rearrange-
ment to more stable cations (Fig. 14.110).
PROBLEM 14.40 When 1-fluorobutane is dissolved in superacid at low tempera-
ture, it is possible to observe a stable species. However, the
13
C NMR spectrum
of that stable species shows only two signals. (a) Why doesn’t the “expected” spec-
trum appear? What is the species that is observed? (b) Although part (a) is quite
easy, this reaction actually poses a vexing problem. Try to write a mechanism for
formation of the stable species. What problems appear when you do?
14.14 Special Topic: Benzyne
In Section 14.12, we mentioned the lack of reactivity of halobenzenes with
nucleophiles. It took the activation of a nitro group on the ring for substitution
to occur (see Figs. 14.94–14.96). In truth, if the nucleophile is basic enough, even
chlorobenzene will react. In the very strongly basic medium potassium amide
(K
NH
2
) in liquid ammonia (NH
3
), chlorobenzene is converted into aniline
(Fig. 14.111).

14.14 Special Topic: Benzyne 681
–
NH
2
Na
+
NH
3
Cl
NH
2
+
NH
2
FIGURE 14.112 A clue to the mechanism for this reaction is provided by Roberts’
observation of isotopic scrambling when labeled chlorobenzene is used.The red
dot represents
14
C, an isotopic label.
The mechanism is surely not an S
N
2 displacement,and there is no reason to think
S
N
Ar addition–elimination is possible with an unstabilized benzene ring.This dif-
ficult mechanistic problem begins to unravel with the observation by John D.Roberts
(b. 1918) and his co-workers that labeled chlorobenzene produces aniline labeled
equally in two positions (Fig. 14.112).
–
NH
2
Na
+
NH
3
Cl
+
NH
2
H
NaCl
..
..
–
(a)
(b)
(a)
(b)
H
NH
2
..
NH
2
H
..
NH
3
NH
2
..
NH
2
..
..
..
–
–
Benzyne
NH
2
..
..
–
+
+
+
+
WEB 3D
FIGURE 14.113 The elimination–addition variation of the S
N
Ar reaction begins with
a halobenzene.The symmetrical intermediate benzyne is formed. Addition of the
amide ion to benzyne produces the observed labeling results.
PROBLEM SOLVING
Almost all “magical migration” reactions, such as the mysterious movement of the
red label in Figure 14.112, involve the formation of a symmetrical intermediate—
here benzyne (Fig. 14.113). The Special Topic in Chapter 21 is full of such
problems, but they have come up elsewhere as well. Problems 14.67, 20.16, and
20.42b are nice examples.
Benzyne formation
What reactions of halides do you know besides displacement? The E1 and E2
reactions compete with displacement reactions (Chapter 7). If HCl were lost
from chlorobenzene, a cyclic alkyne, called benzyne (or dehydrobenzene), would
be formed. This symmetrical bent alkyne might be reactive enough to undergo an
addition reaction with the amide ion.If this were the case, the labeled material must
produce two differently labeled products of addition (Fig. 14.113), because the
NH
2
can attack either carbon of the alkyne.This mechanism is the elimination–addition
version of the S
N
Ar reaction.

682 CHAPTER 14 Substitution Reactions of Aromatic Compounds
+
NaCH
3
O
–
CH
3
CH
3
CH
3
H
3
C
OCH
3
CH
3
OH
(c)
(d)
Benzyne
O
O
(a)
(b)
One would therefore not be too surprised to find that this molecule is able to
add strong bases such as the amide ion. In fact, one would expect very high reactiv-
ity for this strained alkyne. Problem 14.41 gives you a chance to see some of the
reactions of benzyne.
PROBLEM 14.41 Provide arrow formalisms for the formation of these four products
from benzyne:
14.15 Special Topic: Biological Synthesis
of Aromatic Rings; Phenylalanine
Some of the amino acids from which our tissues are constructed contain benzene rings,
and so nature must have a way of making them. You might actually set yourself that
problem right now; you have at your disposal the means to generate benzene from
simple hydrocarbon precursors.
Benzyne is surely an unusual species and deserves a close look.Although this inter-
mediate retains the aromatic sextet, the triple bond is badly bent, and there must be
very severe angle strain indeed.Remember, the optimal angle in triple bonds is 180°.
Moreover, the “third” bond is not composed of 2p/2p overlap like a normal alkyne,
but rather of overlap of two hybrid orbitals (Fig. 14.114).
Benzyne
H
HH
H
=
These π bonds are
the remainder of
the
π system
These orbitals are
part of the
π system
These orbitals are not in the
π system—they make up
the “extra” bond in benzyne
FIGURE 14.114 The structure of
benzyne.
PROBLEM 14.42 Design a synthesis of benzene starting from inorganic com-
pounds, and organic compounds containing only hydrogen and no more than four
carbon atoms. Hint: Your primary goal is to construct the six-membered ring. You
have only one easy way to do that (so far).

14.15 Special Topic: Biological Synthesis of Aromatic Rings; Phenylalanine 683
Nature can’t use the method you designed in Problem 14.42, because too much
energy is needed to do the crucial reaction that constructs the six-membered ring.
Nature starts from simple sugars, which contain the carbons necessary for the
ring, along with many hydroxyl groups. (For much more on carbohydrates, wait for
Chapter 22.) The overall strategy, if one may speak of Nature having a strategy, is
to build a cyclohexane ring, then use the OH groups in various ways to do elimi-
nation reactions and put in the necessary double bonds.
Here is how the process works for phenylalanine, one of your amino acids.
A sugar is transformed enzymatically into dehydroquinate (A), which undergoes an
elimination reaction,also enzyme mediated,to give dehydroshikimate (B) (Fig.14.115).
COO
–
OH
HO
OH
O
COO
–
OH
HO
HO
COO
–
OH
OH
O
A sugar
Dehydroshikimate
(B)
Dehydroquinate
(A)
O
OPO
3
2–
FIGURE 14.115 In the biosynthesis of phenylalanine, enzymes transform a sugar into
dehydroquinate (A) and then dehydroshikimate (B).
The carbon–oxygen double bond in B is reduced and phosphorylated to give
shikimate-3P (C) (Fig. 14.116). Another enzyme adds a three-carbon fragment to
one of the hydroxyl groups (you’ll see why this is critical in a moment) to give D,
which undergoes a second elimination to place the second double bond in the ring
in E, chorismate.
COO
–
COO
–
OH
OH
O
Dehydroshikimate
(B)
COO
–
OH
OH
–2
O
3
PO
Shikimate-3P
(C)
–2
O
3
PO
COO
–
OH
O
Chorismate
(E)
COO
–
O
COO
–
OH
Enolpyruvylshikimate-3P
(D)
FIGURE 14.116 Dehydroshikimate
(B) is transformed into chorismate (E).
So far the strategy we guessed at is working—Nature has effectively done two
elimination reactions and seems poised to do a third. But now the side chain that
was added to make D in Figure 14.116 comes into play, and chorismate mutase

684 CHAPTER 14 Substitution Reactions of Aromatic Compounds
OH
O
O
–
O
NH
3
Prephenate
(F)
Phenylpyruvate
(G)
Phenylalanine
COO
–
COO
–
O
COO
–
FIGURE 14.118 Finally, prephenate
(F) is transformed into
phenylalanine.
The final elimination occurs in prephenate, with the carboxylate (COO
) directly
attached to the ring acting as trigger. Elimination of this group and the hydroxide
leads to phenylpyruvate (G) (Fig. 14.118). Now the three-carbon chain is elaborated
into the side chain of the amino acid phenylalanine.
NH
3
OH
COO
–
Tyrosine
This sequence is far from simple,but even though it is laden with bells and whis-
tles,some of which we surely could not anticipate, the general adherence to our early
strategy—build the six-membered ring and then use the hydroxyls as leaving
groups—is apparent.
PROBLEM 14.44 There is another amino acid, tyrosine, that is formed from
prephenate (Fig. 14.117). At first this transformation seems surprising, perhaps
even impossible. Analyze what must be done and explain why it will not be easy.
induces a transformation of E to prephenate,F (Fig. 14.117).You can see this process
in some detail in Chapter 20.
COO
–
O
OH
COO
–
COO
–
–
OOC
OH
O
enzyme
Chorismate
(E)
Prephenate
(F)
FIGURE 14.117 Chorismate (E) is
made into prephenate (F).
PROBLEM 14.43 Write an arrow formalism for the transformation of compound
E into F. Don’t worry about the role of the enzyme.

14.16 Summary 685
+
(+) (+)
BH
B
E
+
E
..
H
E
–
+
FIGURE 14.119 The general mechanism for electrophilic aromatic substitution.
Key Terms
acid chloride (p. 643)
acylium ion (p. 643)
benzyne (p. 681)
Chichibabin reaction (p. 676)
diazonium ion (p. 647)
electrophilic aromatic substitution
(p. 635)
Friedel–Crafts acylation (p. 643)
Friedel–Crafts alkylation (p. 639)
ipso attack (p. 677)
Meisenheimer complex (p. 675)
nucleophilic aromatic substitution (p. 675)
[n]paracyclophane (p. 628)
Sandmeyer reaction (p. 649)
superacid (p. 680)
Reactions, Mechanisms, and Tools
By far the most important reaction in this chapter is elec-
trophilic aromatic substitution by a variety of electrophiles (E
).
This reaction involves the initial formation of a resonance-
stabilized, but not aromatic, cyclohexadienyl cation.
Deprotonation regenerates an aromatic system (Fig. 14.119).
Nucleophilic aromatic substitution occurs only when
the ring carries substituents that can stabilize the negative
charge introduced through addition of a nucleophile.
Departure of a leaving group regenerates the aromatic system
(Fig. 14.100).
14.16 Summary
New Concepts
This chapter involves the chemistry of aromatic compounds
and is dominated by two fundamental interrelated notions.
First, it is simply difficult to disturb the aromatic sextet, and
second, once aromaticity is disrupted, it is easily regained. In
chemical terms, this means that reactions of benzene and
other simple aromatic compounds generally involve a highly
endothermic first step with formation of a high-energy inter-
mediate. In electrophilic aromatic substitution, the most com-
mon reaction of aromatic compounds, this intermediate is the
cyclohexadienyl cation. Because this intermediate lies far
above benzene in energy, the transition state leading to it is
also high in energy, and the reaction is difficult. Reactions of
the cyclohexadienyl cation that regenerate aromatic systems
are highly exothermic, and therefore have low activation ener-
gy barriers.
Further substitution reactions of an already substituted
benzene are controlled by the properties of the first sub-
stituent. Both resonance and inductive effects are involved, but
the resonance effect usually dominates. Many substituents can
stabilize an intermediate cyclohexadienyl cation through reso-
nance if substitution is in the ortho or para position. The extra
stabilization afforded by the substituent leads to relatively high
rates of reaction. Positively charged substituents generally sub-
stitute meta, because ortho or para substitution places two pos-
itive charges on adjacent positions. The introduction of a
second positive charge means that these substitutions are rela-
tively slow.
At low temperature, in sufficiently polar, but nonnucleo-
philic solvents, some carbocations are stable. Even under
these superacid conditions, rearrangements through hydride
or alkyl shifts to the most stable possible carbocation are
common.

686 CHAPTER 14 Substitution Reactions of Aromatic Compounds
Wolff–Kishner reduction
Δ
O
NH
2
NH
2
/base
ethylene glycol
Clemmensen reduction
Δ
O
Zn/Hg/HCl
R
R
C
R
HH
R
H
H
C
R
R
R
R
R Cl
Friedel–Crafts alkylation; carbocationic
rearrangements occur to give the most
stable ion; the possible structures of
R are therefore limited
AlCl
3
Wolff–Kishner
or
Clemmensen
reduction
R
R
O
R
1. Alkanes
Sn/HCl
or
Reduction of nitro groups
catalytic
hydrogenation
NO
2
NH
2
2. Alkylbenzenes
3. Anilines
4. Benzenes
Reversal of sulfonation reaction
H
2
O/H
3
O
+
Δ
The diazonium ion can be made from nitrobenzene
H
3
PO
2
+
N
2
SO
2
OH
5. Benzenesulfonic Acids
SO
3
HOSO
2
OH
Simple sulfonation
SO
2
OH
6. Cyanobenzenes
Sandmeyer reaction
CuCN
CN
N
2
+
7. Cyclohexanes
High-pressure catalytic hydrogenation
H
2
/Pt
polar solvent, pressure
Syntheses
This chapter provides many new syntheses of aromatic com-
pounds. Although we can now make all sorts of substituted
benzenes, there is still a unifying theme: Each product ultimately
derives from a reaction of benzene with an electrophile.

14.16 Summary 687
8. Deuterated Benzenes
See also methods for making benzenes in item 4
DOSO
2
OD
D
6
9. Diazonium Ions
HONO/HCl
–
Cl
NH
2
N
2
+
10. Halobenzenes
Aromatic halogenation
Aromatic halogenation
Br
2
/AlBr
3
Cl
2
/AlCl
3
Sandmeyer reaction
CuCl
Br
Cl
Cl
N
2
+
Sandmeyer reaction
CuBr
Br
N
2
Schiemann reaction
KI
HBF
4
I
F
N
2
N
2
+
+
11. Nitrobenzenes
CF
3
COOOH
Oxidation of aniline
NO
2
NH
2
Simple nitration
HONO
2
HOSO
2
OH
NO
2
12. Phenols
Δ
H
2
O
OH
N
2
+
13. Phenyl Ketones
AlCl
3
Friedel–Crafts acylation; no rearrangements occu
r
O
C
R
Cl
R
O
C
688 CHAPTER 14 Substitution Reactions of Aromatic Compounds
14.17 Additional Problems
PROBLEM 14.45 Explain why the trifluoromethyl group is
meta-directing in electrophilic aromatic substitution. Would you
expect CF
3
to be activating or deactivating? Why?
CF
3
CF
3
E
E
+
PROBLEM 14.46 Show where the following compounds would
undergo further substitution in electrophilic aromatic substitution.
There may be more than one position for further substitution in
some cases. Explain your reasoning.
NO
2
NH
2
NO
2
CF
3
CH
3
NO
2
NO
2
CH
3
CH
3
NH
2
NO
2
NO
2
OH
CH
3
OH
C
O
CH
3
C
O
H
3
C
(a) (b) (c)
(d)
(e)
(f)
(g)
(h)
O
CH
3
HN
C
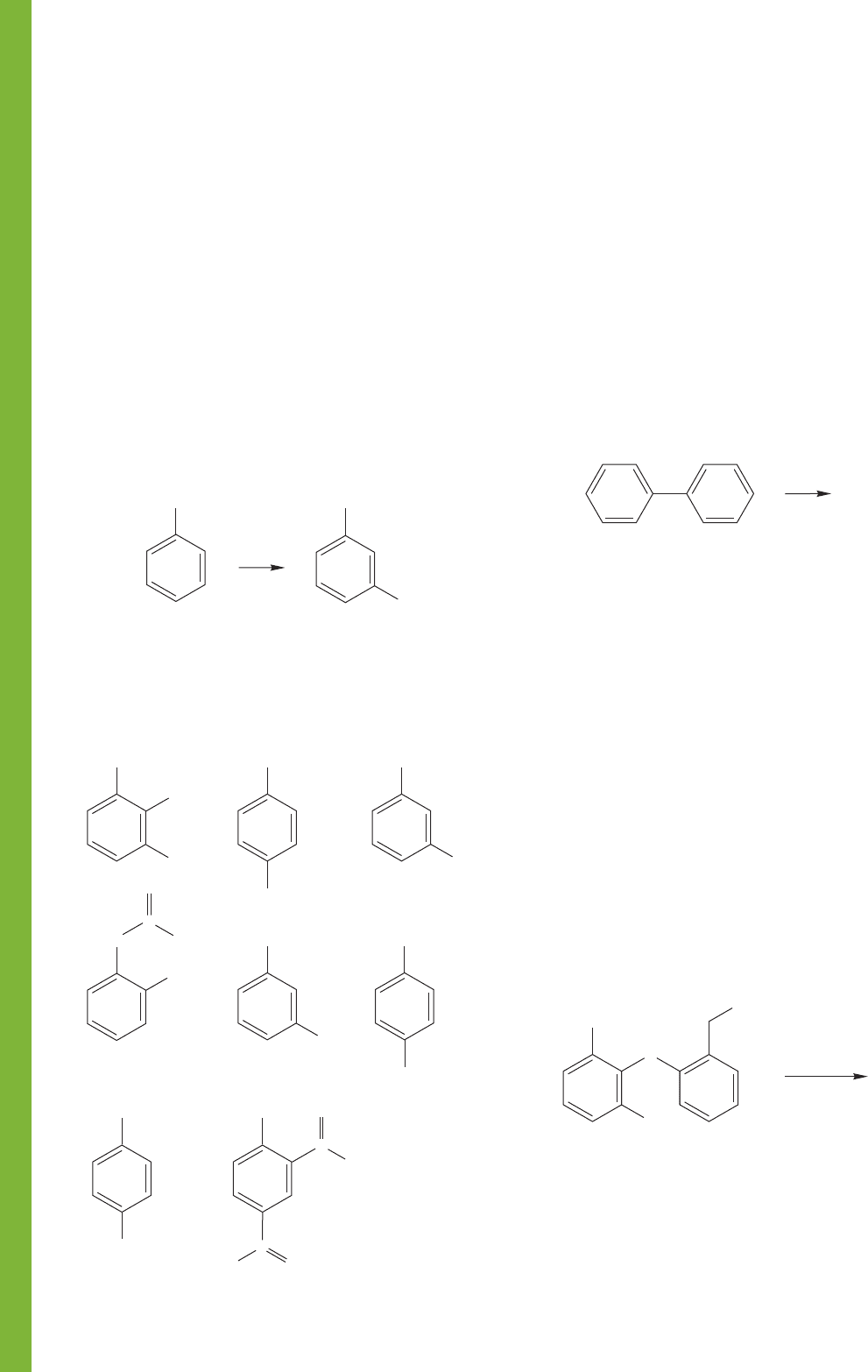
PROBLEM 14.47 Predict the position of further substitution in
biphenyl. Explain your reasoning.
E
+
Biphenyl
?
NO
2
?
+
Cl
Cl
H
N
COOH
Diclofenac
PROBLEM 14.48 Show specific reagent(s) and product(s) for
the following reactions with anisole (methoxybenzene):
(a) nitration
(b) alkylation
(c) sulfonation
(d) monobromination
(e) acylation
PROBLEM 14.49 Show the product(s) for chlorination (Cl
2
,
FeCl
3
) of the following compounds:
(a) toluene
(b) chlorobenzene
(c) 4-nitrotoluene
(d) 2,4-dinitrophenol
(e) diphenylether
PROBLEM 14.50 Diclofenac is a nonsteroidal antiinflammato-
ry drug (NSAID) that is effective for treating pain. In an effort
to better understand its enzyme selectivity, it has been nitrated.
A single product was obtained. Predict the product and explain
your choice.
PROBLEM 14.51 Predict the product for the first reaction shown
on the next page. What mechanism do you suppose is involved
in making the product you have predicted? Based on your
answer to the first reaction, what product do you predict for the
second reaction shown?
Common Errors
The most common conceptual error is to attempt to analyze the
course of aromatic substitution reactions by looking at the start-
ing materials and products. The various resonance and inductive
effects discussed in this chapter are far more important in the
charged intermediates and in the transition states leading to
those intermediates. When analyzing one of these reactions,
always look first at the cyclohexadienyl intermediate (a cation
in electrophilic aromatic substitution and an anion in the less
common nucleophilic reactions), and then consider the transi-
tion state leading to it.
Another potential error is failing to keep up with the prolif-
erating synthetic detail. Be sure to look at each new reaction in
this and subsequent chapters with an eye to using it in synthesis.
Keep up your file cards!