Claverie J-M., Notredame C. Bioinformatics for Dummies
Подождите немного. Документ загружается.


The next form that appears (titled “Formatting BLAST”) allows you to
change a number of output features. (See Figure 2-26.) For now, keep the
defaults as they are.
7. Click the Format! button.
BLAST now gives you an estimated running time for your query. If the
time of the day is right, this shouldn’t take more than a few seconds.
8. When the Results page appears, scroll down the page until you reach
a long list of sequences (Figure 2-27).
What you have here are all the sequences that have a significant similar-
ity with your query sequence, ranked by decreasing score values. If you
used
DUT_ADEG1 or another protein sequence taken from a database, the
best matching protein is probably the one you started with. (This is what
happened in Figure 2-27, with the Fowl adenovirus dUTPase.) Here the
highest similarity score is 361. The score value depends on the length of
the most-similar segments that occur between two sequences — as well
as on the quality of the match. It is not normalized to 100 percent. (See
Chapter 7 to find out more about the significance of BLAST scores.)
In this list of sequences, you would simply click an underlined identifier
to link to the corresponding
nr (nonredundant) database entry.
9. Click one of the underlined scores in the second column from the right.
This brings you further down in the page (Figure 2-28), to see the align-
ment between your query sequence and the matching nr sequence of
the protein that corresponds to this score.
In our example, the best match is (not surprisingly) an identical one. By going
down the list, you can see less-than-perfect matches, slowly degrading as the
corresponding score decreases and the E-value increases. The
E-value is an
Click here once and wait.
Figure 2-26:
NCBI
BLAST
“format-
while-you-
wait” form.
60
Part I: Getting Started in Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 60

assessment of the statistical significance of the score. E-values close to 1 are
a warning that the conclusion you might draw from the alignments is not reli-
able. (Go to Chapter 7 to find out more about these issues.)
Query sequence
Database sequence
Matching sequence
Figure 2-28:
NCBI
BLAST
output form:
local
sequence
alignments.
Click here to see the nr entry.
Click here to see the corresponding alignment.
Figure 2-27:
NCBI
BLAST
output form:
best match
listing.
61
Chapter 2: How Most People Use Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 61

Making a Multiple Protein Sequence
Alignment with ClustalW
Besides running database searches to identify similar proteins pair by pair,
the second most common bioinformatic task that biologists like to perform
with protein sequences is a multiple alignment.
Multiple alignments consist in
lining up many similar proteins side by side for the sake of comparison.
Multiple alignments are used to
Identify sequence positions where specific amino acids really matter for
the structural integrity or the function of a given protein
Define specific sequence signatures for protein families
Classify sequences and build evolutionary trees
We detail all the intricacies of making good multiple alignments in Chapter
9. At this point, however, we simply want to show you that performing a
multiple alignment is really easy — especially when you have the pleasure
of using some nice Internet server, such as the one maintained by the Protein
Information Resource (PIR) people at Washington, D.C.’s Georgetown
University.
The PIR actually originated from the
Atlas of Protein Sequences, the first pro-
tein-sequence collection (which was built by the late Prof. M. Dayhoff in the
late 1970s). The PIR site offers some useful protein analysis tools and data-
bases that we invite you to explore by yourself. Among these tools, it offers a
multiple-alignment server (running the standard ClustalW program) that is
really easy to use for beginners.
Do the following to get your feet wet using ClustalW:
1. Point your browser to pir.georgetown.edu.
The PIR home page appears, as shown in Figure 2-29.
2. Under the Search/Analysis heading, choose Multiple Alignment from
the drop-down menu to display the input form.
The input form appears. At this point, you need a few FASTA-formatted
protein sequences.
3. Open the dUTPase FASTA-formatted sequence file that you created on
your PC in the previous section of this chapter.
62
Part I: Getting Started in Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 62

Alternatively, you can go to the Swiss-Prot home page at
www.expasy.org/sprot/
and grab a few dUTPase sequences (such as DUT_AQUAE, DUT_BRAJA,
DUT_BPT5, DUT_CANAL, and DUT_BUCAI), as we spell out in the section
“ExPASy: A prime Internet site for protein information,” earlier in this
chapter.
4. Copy/paste five of these sequences (all at once) into the input window,
as shown in Figure 2-30.
Be careful to copy the definition line and the entire amino-acid sequence
for each of them.
5. Click the Submit button.
Within a few seconds, your screen (Figure 2-31) is proudly displaying its
first successful multiple-sequence alignment — and a tree-like represen-
tation of the pair-wise similarity of these sequences.
Select Multiple Alignment.
Figure 2-29:
Another
useful
protein
analysis
center: PIR.
63
Chapter 2: How Most People Use Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 63

This output (if you used the same sequences as we did here) illustrates the
power of multiple alignments:
It clearly delineates a region of high-versus-low sequence similarity in
these related proteins.
Positions 100 percent identical (highlighted by
<*>) or occupied by
chemically similar amino-acids (highlighted by
<:> or <.>) tend to occur
in a clustered fashion along the sequence, defining regions that are prob-
ably directly involved in shaping the catalytic site or performing the
pyrophosphatase biochemical reaction.
Various types of functional signatures (small sequence segments
associated to a given biochemical function) can be extracted from
such a multiple alignment.
Evolutionary relationships between proteins are inferred from
phylogenetic trees.
In Chapters 9, 11, and 13, we tell you more about these key applications of
bioinformatics.
Paste your sequences here.
Click here to start the multiple alignment.
Figure 2-30:
Filling up
the multiple
alignment
input
window
at PIR.
64
Part I: Getting Started in Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 64
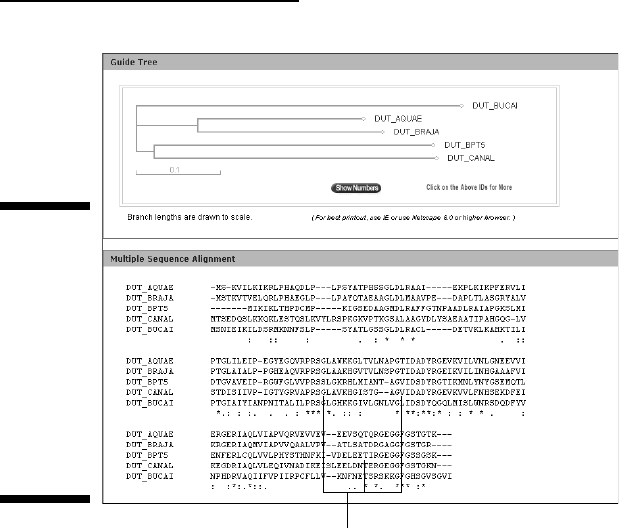
Invariant regions
Figure 2-31:
Multiple
alignment of
five bacterial
dUTPase
sequences
and the
correspond-
ing tree built
from their
pair-wise
similarity.
65
Chapter 2: How Most People Use Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 65

66
Part I: Getting Started in Bioinformatics
06_089857 ch02.qxp 11/6/06 3:52 PM Page 66

Part II
A Survival Guide
to Bioinformatics
07_089857 pp02.qxp 11/14/06 12:10 PM Page 67

In this part . . .
B
ioinformatics is about finding and interpreting bio-
logical data online. In this part, we show you how to
go shopping in the main bioinformatics databases. We’ve
got lots of great tips for picking up the freshest, ripest
data (and not just the low-hanging fruits!). Because all
these databases are linked with one another, we also show
you how you can travel across them and gather every
piece of information you need. It’s a fascinating journey
across human knowledge –– and a compulsory starting
point for any research project.
With the right data in the fridge, it’s time to start cooking.
In this part, we show simple recipes for working with DNA
and protein sequences in order to predict their most basic
properties.
07_089857 pp02.qxp 11/14/06 12:10 PM Page 68

Chapter 3
Using Nucleotide
Sequence Databases
In This Chapter
Nabbing a quick refresher on the structure of genes and genomes
Making use and sense of GenBank
Finding out about a specific gene
Working with complete microbial genomes
Browsing the human and other animal genomes
The secret of success is to know something nobody else knows.
— Aristotle Onassis (1906–1975)
S
equence databases are great tools because they offer a unique window
on the past. They make it possible to answer today’s biological questions
by enabling us to analyze sequences that may have been determined as many
as 25 years ago, when the whole technology emerged. By doing this, they
connect past and present molecular biology.
The first databases were in fact created as some sort of sequence museum,
where sequences could be preserved for all eternity in pristine form, just as
they were determined, interpreted, and published by their original authors.
This
historical (time capsule!) perspective pretty much remains in GenBank,
the leading nucleotide sequence repository maintained as a consortium
between the U.S. National Center for Biotechnology Information (NCBI),
the European Molecular Biology Laboratory (EMBL), and the DNA Data
Bank of Japan (DDBJ). In this chapter, we show you how to use GenBank
and decipher its entries.
Repository-type databases are great tools when you want to come up with a
bibliography for a particular sequence, but they do not provide easy access
to sequence data when your query deals with broader issues related to a
gene or function rather than with a specific paper. For this reason, the
08_089857 ch03.qxp 11/6/06 3:54 PM Page 69