Mitin V.V., Sementsov D.I., Vagidov N.Z. Quantum mechanics for nanostructures
Подождите немного. Документ загружается.


C.3 Degenerate electron gas 393
of occupation of levels with such energy is greater than 1/2. If E > E
F
,then f (E) < 1/2.
Therefore, at T > 0 K levels that are lower than the Fermi level have higher probabilities
of being occupied than do the levels higher than the Fermi level. The Fermi function,
f (E), is practically equal to unity for energies lower than the Fermi energy by several
k
B
T ,whereas f (E) is practically equal to zero for energies higher than the Fermi energy
by several k
B
T . Figure 7.3 shows the Fermi function for two temperatures. The higher
the temperature, the wider is the energy interval over which the Fermi function changes
from f (E) = 1to f (E) = 0. The width of this interval is about (4–6) k
B
T (at room
temperature k
B
T ≈ 0.025 eV). The width of the plot along the energy axis depends
on the magnitude of the Fermi energy, E
F
, which for metals is about several electron-
volts. Thus, the graphical plot of the Fermi function in Fig. 7.3 is merely indicative
since it does not take into account the real width of the function f (E) along the energy
axis.
C.3.2 The Fermi energy and average energy of a
degenerate electron gas
In order to define E
F
let us take into account that this is the energy of the highest level
occupied by electrons at T = 0K.AllN conduction electrons have energy within the
interval from 0 to E
F
. Therefore,
N =
E
F
0
g(E)dE. (C.29)
Here, the function g(E) is the density of states of a three-dimensional crystal, which takes
into account that each energy level can be occupied by two electrons. Let us substitute
into Eq. (C.29) the equation obtained in Section 7.1, namely Eq. (7.16), for the density of
quantum states for three-dimensional crystals:
N =
√
2Vm
3/2
e
π
2
h
-
3
E
F
0
√
E dE =
V (2m
e
E
F
)
3/2
3π
2
h
-
3
. (C.30)
From this expression we get the relationship that connects the concentration of free
electrons, n = N /V , and the Fermi energy, E
F
:
n =
(2m
e
E
F
)
3/2
3π
2
h
-
3
. (C.31)
Equation (C.31) gives an important definition of the Fermi energy, E
F
, in terms of the
concentration of free electrons, n:
E
F
=
h
-
2
2m
e
3π
2
n
2/3
. (C.32)
As an example, let us find the Fermi energy for free electrons in copper, Cu. Since each
copper atom provides one conduction electron (valence electron), the concentration of

394 Appendix C. Crystals as atomic lattices
Table C.2. Fermi-energy values for some metals
Metal Cs K Na Li Ag Al Be Cu
E
F
(eV) 1.53 2.14 3.12 4.72 5.5 11.9 14.6 6.9
free electrons in the Cu crystal is equal to the concentration of Cu atoms:
n =
N
A
ρ
A
=
6.02 ×10
26
× 8.9 ×10
3
64
≈ 8.4 × 10
28
m
−3
, (C.33)
where N
A
is the Avogadro number, ρ is the density of copper, and A is the mass of
one kilomole of copper. On substituting the obtained concentration of free electrons into
Eq. (C.33), we obtain
E
F
=
(1.05 × 10
−34
)
2
2 ×9.1 ×10
−31
3 × π
2
× 8.4 × 10
28
2/3
≈ 1.1 × 10
−18
J ≈ 6.9eV. (C.34)
In Table C.2 we present the Fermi energy of degenerate electron gases in some metals.
Thus, at absolute zero temperature in the allowed energy band in which all of the
valence electrons of a metal are present (this band is called the conduction band), all
levels up to the Fermi level are occupied. At non-zero temperatures the energy of lattice
thermal oscillations is two orders of magnitude less than the Fermi energy. Therefore, when
the temperature of the metal increases, only a small number of electrons that obtained
additional energy from lattice thermal oscillations (as a result of electron scattering
on phonons) will move to higher unoccupied energy levels of the conduction band. Only
electrons that are close to the Fermi level (i.e., electrons that are separated from unoccupied
states by several k
B
T ) can make such a transition. Thus, the change in temperature affects
the energy only of those electrons in the conduction band of the metal which are close to
the Fermi level.
Only a small number of electrons that occupy levels lower than the Fermi level will
move to the unoccupied levels that are higher than the Fermi level. The number of thermally
excited electrons can be estimated with high precision as
N
T
≈ N
2k
B
T
E
F
, (C.35)
where N is the total number of electrons in the conduction band.
The main indication of degeneracy of an electron gas is the fact that the energy
of the whole ensemble and of the individual electrons practically does not depend on
temperature. The electron gas stays degenerate until each of its electrons could exchange
energy with the crystalline lattice. This is possible only when the average energy of lattice
thermal oscillations, k
B
T , is comparable to the Fermi energy, E
F
. The temperature
T
F
=
E
F
k
B
, (C.36)

C.3 Degenerate electron gas 395
at which an electron gas transforms from a non-degenerate state to a degenerate state is
called the degeneracy temperature. In metals at all practically possible temperatures (for
which the metal stays in a condensed state) the electron gas is degenerate. The electron
gas stays non-degenerate only in semiconductors with a concentration of electrons less
than n = 10
19
cm
−3
.
To find the average energy of an electron in a degenerate electron gas at T = 0K,let
us first find the total energy of the electron gas:
E
N
=
E
F
0
Eg(E)dE =
√
2Vm
3/2
e
π
2
h
-
3
E
F
0
E
√
E dE =
V (2m
e
)
3/2
π
2
h
-
3
(E
F
)
5/2
. (C.37)
Let us divide this energy by the number of electrons in the ensemble and take into account
the expression for the Fermi energy (C.32). As a result, we find the following important
relation for the average energy of a single electron:
E
=
E
N
N
=
(2m
e
)
3/2
5π
2
nh
-
3
(E
F
)
5/2
=
3
5
E
F
. (C.38)
From Eq. (C.38) it follows that an electron gas in equilibrium has more electrons with
high energy than electrons with low energy. According to Eq. (C.38), the average energy
of electrons in Cu is close to 4.15 eV.
More accurate consideration shows that the Fermi energy and average electron energy
in a metal change with temperature. With increasing temperature the Fermi energy neg-
ligibly decreases and the average electron energy slightly increases. Thus, when the
temperature increases from 0 K to 1000 K, the Fermi energy of silver, which is equal to
5.5 eV at T =0 K, decreases by only 0.01 eV.
Example C.2. Find the molar heat capacity of an electron gas and the total heat capacity
of a metallic crystal.
Reasoning. According to classical theory, all conduction electrons in a metal must have
input to the heat capacity of the electron gas, as in the case of a monatomic ideal gas.
For one mole of monatomic ideal gas (the number of particles is equal to the Avogadro
number, N
A
), which is at temperature T , the total thermal energy is equal to
E
µ
=
3
2
k
B
TN
A
=
3
2
RT, (C.39)
where R is the universal gas constant. The molar heat capacity of such a gas is defined as
c
µ
=
dE
µ
dT
=
3
2
R. (C.40)
If conduction electrons in metals obey classical statistics, the total molar heat capacity of
a univalent metallic crystal (taking into account the heat capacity of the crystalline lattice
c
cr
= 3R) must be equal to
c
met
= c
cr
+ c
el
= 3R +
3
2
R =
9
2
R. (C.41)
396 Appendix C. Crystals as atomic lattices
However, at room temperature, for most metals the Dulong–Petit law is valid with very
high precision, i.e., the molar heat capacity of a metal (or non-metal) is equal to c
met
= 3R.
This fact shows that the electron gas in metals is not classical, i.e., this gas does not obey
classical statistics, namely Maxwell–Boltzmann statistics.
As we have already mentioned, in metals a change of temperature leads to a change
in energy only of those electrons which are on levels that differ from the Fermi energy by
several k
B
T . Therefore, most of the conduction electrons will not have input to the heat
capacity since their energy stays constant. We can estimate the magnitude of the electron
heat capacity if we take into account that the number of “active” electrons is defined by
N
T
and their energy at thermal excitation increases approximately by 2k
B
T . The energy
of one mole of electron gas can be presented as
E
µ
=
3
5
E
F
N
A
+
3
2
k
B
T
2k
B
T
E
F
N
A
. (C.42)
As a result, an approximate expression for the molar heat capacity of a degenerate electron
gas, c
el
= dE
µ
/dT , has the following form:
c
el
= 6R
k
B
T
E
F
. (C.43)
If each atom of metal has not one but Z valence electrons, then we have
c
el
= 6ZR
k
B
T
E
F
. (C.44)
More accurate calculations of the molar heat capacity of a degenerate electron gas instead
of the coefficient 6 give π
2
/2.
From the expressions written above it follows that the input from conduction electrons
to the total heat capacity of metals at room temperatures is less than 1% of the input from
the lattice heat capacity, which explains the validity of the Dulong–Petit law for metals.
This input becomes noticeable only at very low temperatures, for which the heat capacity
of the crystalline lattice, which is proportional to T
3
, becomes smaller than the electron
heat capacity, which is proportional to T . This temperature range is at liquid-helium
temperatures for most metals.
C.4 Waves in a crystalline lattice and normal coordinates
The kinetic and potential energy of oscillatory motion of atoms in a crystalline lattice are
functions of the displacement of each atom of a crystal. Since the displacements of all
atoms are coupled, the total energy of a lattice, which is the sum of its kinetic and potential
energies, depends on the coupled displacements of the lattice atoms. It is very important
that, for the theoretical analysis and explanation of processes taking place in a crystal, the
total energy of oscillations of atoms during wave propagation is presented in the form of
a sum of energies of independent waves. Such a representation is possible as a result of
transition from the usual representation of displacements of atoms to the so-called normal
coordinates and to the corresponding generalized momenta. Let us introduce them using
the example of a one-dimensional (linear) chain of atoms.

C.4 Waves in a crystalline lattice and normal coordinates 397
Each nth atom of a linear chain participates in an infinite number of displacements
u
n
(q) with different wavenumbers q and corresponding ω
q
and A
q
:
u
n
(q) = A
q
e
i(qan−ω
q
t)
. (C.45)
Arbitrary motion can be presented in the form of the linear superposition of all possible
waves of the form (C.45) with different wavenumbers q and corresponding frequencies
ω
q
and amplitudes A
q
:
u
n
=
q
A
q
e
i(qan−ω
q
t)
+ A
∗
q
e
−i(qan−ω
q
t)
. (C.46)
Let us introduce a set of variables that can be convenient for further analysis:
a
q
=
√
NA
q
e
−iω
q
t
, (C.47)
through which Eq. (C.46) can be rewritten in the form
u
n
=
1
√
N
q
a
q
e
iqan
+ a
∗
q
e
−iqan
. (C.48)
Since the lattice contains a finite number of atoms, N , and from the cyclic boundary con-
ditions it follows that q has N discrete values, q = [2π/(aN)]g,whereg = 1, 2,...,N ,
the summation in Eq. (C.48) can be done over all mentioned values of wavenumber, q.
The kinetic and potential energies of a linear chain of interacting atoms are given by
the expressions
K =
m
2
N
n=1
du
n
dt
2
, (C.49)
U =
β
2
N
n=1
u
n
− u
n−1
2
. (C.50)
From the expression for the potential energy (C.50) we can obtain the force acting on the
nth atom from the neighboring atoms. Indeed,
F
n
=−
∂U
∂u
n
=−β
2u
n
− u
n−1
− u
n+1
. (C.51)
On substituting Eq. (C.48) into the expressions for energies of Eqs. (C.49)and(C.50)and
taking into account that
da
q
dt
=−iωa
q
,
da
∗
q
dt
= iωa
∗
q
, (C.52)
we come, after some cumbersome transformations, which we will omit here (see Example
C.3), to the following expressions:
K =
m
2
q
ω
2
q
2a
q
a
∗
q
− a
q
a
−q
− a
∗
q
a
∗
−q
, (C.53)
U =
m
2
q
ω
2
q
2a
q
a
∗
q
+ a
q
a
−q
+ a
∗
q
a
∗
−q
. (C.54)

398 Appendix C. Crystals as atomic lattices
The total energy of a linear chain reduces to
E = K + U = 2m
q
ω
2
q
a
q
a
∗
q
. (C.55)
Let us define now the normal coordinates of the lattice,
x
q
= a
q
+ a
∗
q
, (C.56)
and the corresponding momenta, which, taking into account Eq. (C.52), have the form
p
q
= m
dx
q
dt
=
mω
q
i
a
q
− a
∗
q
. (C.57)
Both of the quantities x
q
and p
q
are real. Let us evaluate the parameters a
q
and a
∗
q
:
a
q
=
1
2
x
q
+ i
p
q
mω
q
, a
∗
q
=
1
2
x
q
− i
p
q
mω
q
, (C.58)
and let us substitute them into the expression for the total energy (C.55). As a result we
obtain
E =
q
p
2
q
2m
+
mω
2
q
x
2
q
2
. (C.59)
It follows that, represented in normal coordinates and corresponding momenta, the total
energy of the wave motion of atoms in a linear chain is the sum of energies of independent
harmonic oscillators, h
-
ω
q
. This representation, as will be shown in the next sections,
allows us to describe the interaction of electrons in a crystal with lattice vibrations
using the language of crystal quasiparticles. In addition, the elementary lattice excitations
represented in terms of the normal coordinates (Eq. (C.59)) are called phonons. Phonons
are one of the main types of quasiparticle in a crystal.
Example C.3. Obtain the expression for the kinetic energy of oscillations of atoms in a
linear chain using the representation of an atom’s displacement Eq. (C.48) expressed in
terms of the variables a
q
introduced by the relationship (C.47).
Reasoning. Let us substitute Eq. (C.48) into the expression for the kinetic energy (C.49):
K =
m
2
N
n=1
du
n
dt
du
n
dt
. (C.60)
Taking into account Eq. (C.48),
K =
m
2N
N
n=1
d
dt
q
a
q
e
iqan
+ a
∗
q
e
−iqan
d
dt
q
a
q
e
iq
an
+ a
∗
q
e
−iq
an
. (C.61)
C.4 Waves in a crystalline lattice and normal coordinates 399
In the above equations only a
q
and a
q
are functions of t; thus, taking into account
Eq. (C.52), we get
K =
m
2N
qq
N
n=1
ω
q
ω
q
×
a
q
a
q
e
i(q+q
)an
− a
q
a
∗
q
e
i(q−q
)an
− a
∗
q
a
q
e
−i(q−q
)an
+ a
∗
q
a
∗
q
e
−i(q+q
)an
. (C.62)
Let us do the summation in the last expression over the index n first, using the fact that
the sums over this index are geometric progressions. Indeed,
N
n=1
e
i(q±q
)an
=
N
n=1
e
i
2π
N
gn
= e
i
2π
N
g
+ e
i
2π
N
2g
+···+e
i
2π
N
Ng
=
e
i
2π
N
g
1 −e
i2πg
1 − e
i
2π
N
g
=
0, g = 0, q ±q
= 0,
N, g = 0, q ± q
= 0,
(C.63)
where we must take into account that the wavenumber q + q
, reduced to the first Brillouin
zone, takes N values because the number g = 1, 2,...,N . Thus, the above-mentioned
sum plays the role of Kronecker’s symbol, i.e.,
1
N
N
n=1
e
i(q±q
)an
= δ
q±q
=
0, q ± q
= 0,
1, q ± q
= 0,
(C.64)
which allows us to do the summation over the index n in Eq. (C.62):
K =
m
2
qq
ω
q
ω
q
a
q
a
q
δ
q+q
− a
q
a
∗
q
δ
q−q
− a
∗
q
a
q
δ
q−q
+ a
∗
q
a
∗
q
δ
q+q
. (C.65)
Taking into account that ω
−q
= ω
q
as a result of summation over the index q
, we come
to Eq. (C.53).
Example C.4. Find the molar heat capacity of a crystalline lattice consisting of atoms of
the same type, using the classical description of the energy of thermal vibrations.
Reasoning. The crystalline lattice contains N atoms, which are in a state of thermo-
dynamic equilibrium at a given temperature T . Atoms oscillate near their positions of
equilibrium. For an arbitrary atom of the three-dimensional crystal there are three degrees
of freedom of oscillatory motion (oscillatory motion in three independent directions).
Considering a crystal as a system of non-interacting three-dimensional oscillators, we can
write the crystal’s energy of thermal motion as
E(T ) = 3N
ε(T )
, (C.66)
where
ε(T )
is the mean thermal energy of a single linear oscillator that depends on the
temperature. To find it, let us take into account that the mechanical energy of a harmonic

400 Appendix C. Crystals as atomic lattices
oscillator averaged over a single period (or over a big time interval) is, according to
Eq. (A.40), equal to
ε = K +U = 2K . (C.67)
Here we took into account that the mean potential and kinetic energies of an oscillator are
equal to each other. Thus,
E(T ) = 6N
K (T )
. (C.68)
In accordance with the well-known theorem of classical physics, in thermodynamical
equilibrium each degree of freedom of a particle’s translational motion corresponds to the
mean kinetic energy of the crystal’s thermal vibrations:
K (T )
= k
B
T/2. Thus, for the
total energy of the crystal’s thermal vibrations we obtain
E(T ) = 3Nk
B
T. (C.69)
If the number of atoms in a crystal is equal to the Avogadro number, N
A
, i.e., if we take
one mole of a substance, its energy will be
E(T ) = 3N
A
k
B
T = 3RT, (C.70)
where R = N
A
k
B
= 8.31 J mol
−1
K
−1
is the so-called universal gas constant. Since the
energy found is an internal energy of a crystal, then, by definition, the molar heat capacity
is equal to
c =
dE
dT
= 3R. (C.71)
This relation is called the Dulong–Petit law. It works sufficiently well at high temperatures
(T > T
D
,whereT
D
is the so-called Debye temperature of the given substance), but it is not
valid for low temperatures. At T → 0 the molar heat capacity of all solids tends to zero.
This fact can be explained only with the help of the quantum-mechanical description. If
the substance is complex and in a unit cell there are s different atoms or ions, then its
molar heat capacity is defined by the relation
c = 3sR. (C.72)
C.5 The energy spectrum of an electron in a crystal
As we have already mentioned, ordinary crystals, which have as a structural unit individual
atoms or ions and distances between them of several
˚
angstr
¨
om units, are superlattices with
various types of ordering of their structural units. The correct description of the electron
dynamics in such a superlattice, i.e., in a periodic field of atomic nuclei, requires a
quantum-mechanical approach based on the solution of the Schr
¨
odinger equation. The
behavior of the entire system of electrons and nuclei is defined by its wavefunction.
In our case, even without taking into account spin, the wavefunction of such a system

C.5 The energy spectrum of an electron in a crystal 401
will depend on the 3N spatial coordinates of the nuclei and the 3ZN coordinates of the
electrons (here Z is the number of electrons per atom). Since for a crystal of macroscopic
size N is approximately equal to 10
23
, finding such a wavefunction and its analysis must
be understood to be unrealistic. Because of this, for the solution of problems involving
electrons in crystals certain well-established approximations, which allow one to calculate
physical quantities observed in experiments, are used. The main approximations that are
used in the electron theory of crystals are the adiabatic and one-electron approximations.
In the adiabatic approximation, which is based on the smallness of the ratio of the
electron mass to the mass of a nucleus, the quantum-mechanical problem of the behavior
of the system of electrons and nuclei splits into two simpler problems. The first problem
is connected with finding the state of the electrons in the field of motionless nuclei, which
are located at the vertexes of the crystalline lattice, i.e., at their equilibrium positions.
The second problem is connected with the consideration of small-amplitude oscillations
of nuclei near their positions of equilibrium as a result of interaction with the system
of electrons whose states were defined in the first stage. This problem is reduced on the
classical level to studying elastic oscillations and waves in crystalline structures. The
correctness of the classical approach for the consideration of lattice vibrations is justified
by the smallness of the de Broglie wavelength of the atoms and ions in comparison with
the lattice constant (λ
Br
a). In the case of thermal oscillations of a crystalline lattice
the velocity of oscillating atoms may be estimated as follows:
v
T
≈
3k
B
T
m
. (C.73)
Using Eq. (C.73), we can find the de Broglie wavelength of these atoms:
λ
Br
=
2π h
-
mv
T
≈
2π h
-
√
3mk
B
T
. (C.74)
Since for Si atoms m ≈ 4.7 ×10
−26
kg, their de Broglie wavelength is equal to λ
Br
≈
3 ×10
−11
m, which is one order of magnitude smaller than the lattice constant, a.
The one-electron approximation allows significant simplification of the complex prob-
lem of description of the motion of a system of electrons interacting with each other and
with the motionless nuclei. This problem is reduced to the simpler problem of independent
motion of each electron in a self-consistent periodic field, U (r), which is created by all
other particles of the crystalline structure. The form of this field is defined by the symme-
try properties of the crystal. Even though the coordinate dependence of U(r) is almost the
same for different crystals, there are sharp differences among the behaviors of electrons
in metals, dielectrics, and semiconductors. To understand the origin of these differences,
let us analyze the behavior of electron systems in crystals using the above-mentioned
approximations.
C.5.1 The wavefunction of an electron in a crystal
Considering hydrogen molecules in Section 6.6, we showed that electron atomic states
become non-stationary when atoms form a molecule. Owing to the tunneling phenomenon,
electrons in a molecule constantly tunnel from one atom to another, thus becoming
402 Appendix C. Crystals as atomic lattices
Na
E
2
E
1
E
2
E
1
0
a
2a
2a
x
U
E
2
E
1
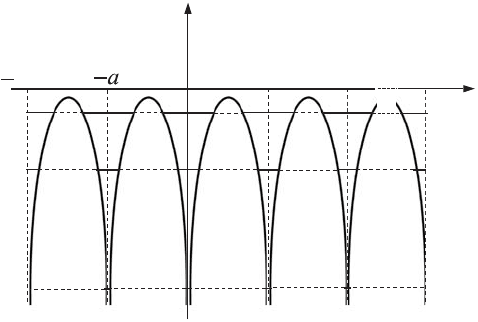
Figure C.9
Inner-crystalline potential.
collectivized for both atoms. This collectivization provides strong covalent bonding. An
analogous situation takes place in a crystal, whose potential energy has the form of
regularly placed potential wells separated by potential barriers. Figure C.9 shows a one-
dimensional case of such a distribution of potential energy. Each electron of any atom
in a crystal has a limited probability of tunneling through the potential barrier from one
atom to another. The maximum probability will be for transitions between the same states
of electrons in atoms. Since the probability of tunneling through the barrier decreases
exponentially with increasing width and height of the potential barrier, electrons from the
outermost shells have a higher probability of tunneling than do the internal electrons.
When two hydrogen atoms unite to form a hydrogen molecule, as a result of the
Pauli exclusion principle each atomic level, E
q
(the index q defines the set of all electron
quantum numbers in an atom) splits into two close sublevels (see Section 6.6). The distance
between two sublevels is defined as
E
(2)
q
− E
(1)
q
≈ 2 A
q
E
q
, (C.75)
where A
q
is the exchange integral for corresponding atomic states. An increase of the
number of atoms binding increases the number of sublevels into which each initial atomic
level is split. As a result, because of the exchange interaction, each atomic level, defined
by a set of quantum numbers {q}, transforms into a band of closely placed sublevels whose
width is proportional to 2A
q
. For deep-lying atomic levels, which correspond to internal
electrons, this splitting is small. Therefore, these deep-lying bands are narrow.
For levels that correspond to the electrons from the outermost shells the splitting
is considerable and the width of bands may be comparable to the separation between
atomic levels. The electron quantum states of an individual atom are not in general
stationary for a crystal. Each electron, as a result of tunneling from atom, to atom,
belongs not to an individual atom but to the entire crystal. Only the internal electrons can
be considered as belonging to their corresponding atoms. To describe such electrons in a
crystal, their wavefunctions, taking into account the degeneracy of states, can be presented
as a superposition of atomic wavefunctions ψ
0q
(r −a
n
), corresponding to the vertex a
n