Westermeier R., Naven T., H?pker H.-R. Proteomics in Practice: A Guide to Successful Experimental Design
Подождите немного. Документ загружается.

3 Mass Spectrometry240
a)
b)
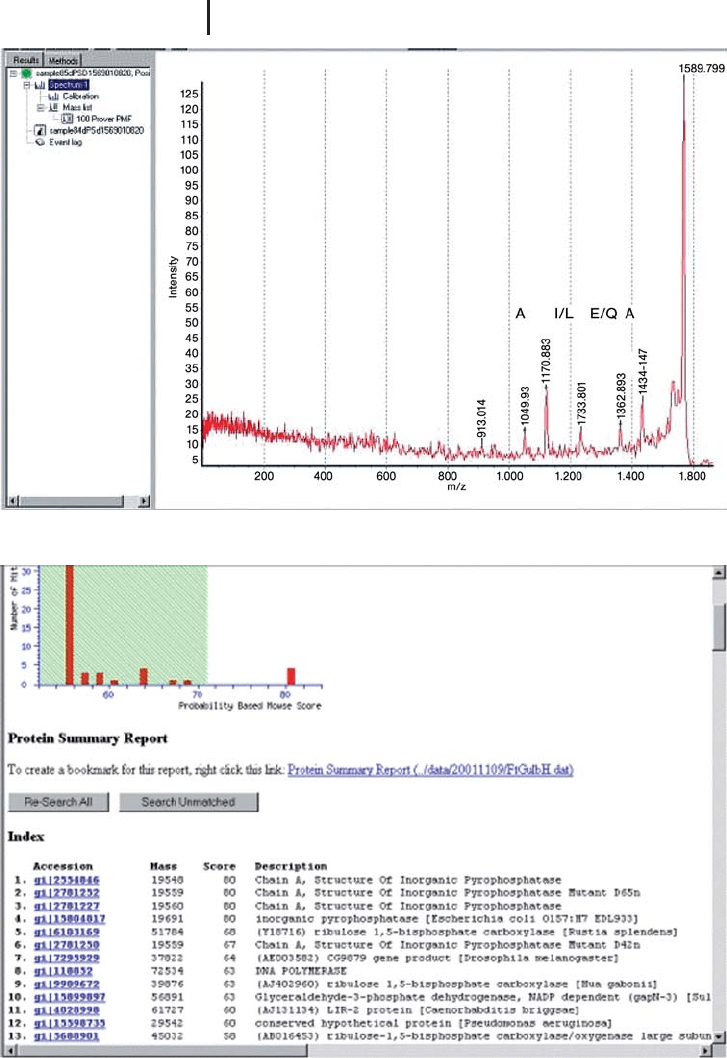
Fig. 3.17: (a) PSD spectrum of the derivatized peptide, m/z 1569.79. The sequence – A[IL]EA
was determined from the spectrum and combined with the PMF as before. The result of the
combined search is demonstrated in Figure 3.17b. An unambiguous result is returned from the
search. (b) MASCOT search result from the combined PMF and partial sequence determined
from the PSD spectrum in Figure 3.17a.
3.3 Generating MS Data for Protein Identification 241
a)
b)

3 Mass Spectrometry242
c)
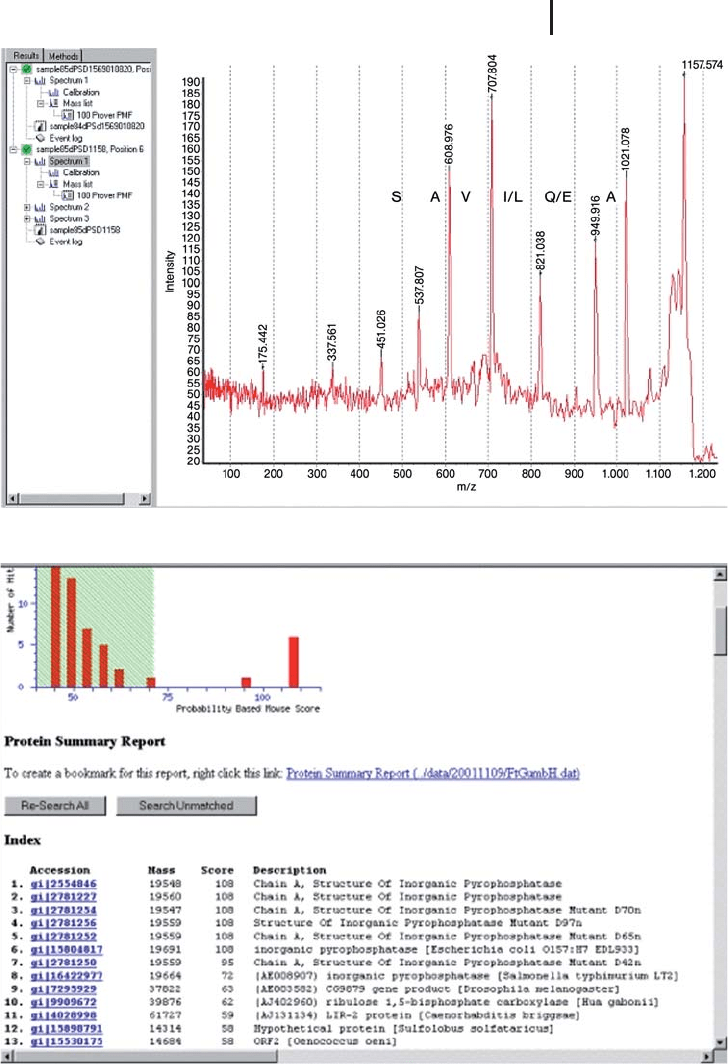
Fig. 3.18: (a) PSD spectrum of the derivatized peptide, m/z 1157.57.
The sequence – SAV[IL]EA was determined from the spectrum and
combined with the PMF as before. The result of the combined search
is demonstrated in Figure 3.18b. An unambiguous result is returned
from the search. (b) MASCOT search result from the combined PMF
and partial sequence determined from the PSD spectrum in Figure 3.17a.
(c) MASCOT search result of the combined PMF and partial sequence
from two peptides.
3.3.4
Tandem Mass Spectrometry
Tandem mass spectrometry (or MS/MS) is performed with a certain
type of mass spectrometer. Such an instrument needs to be capable
of selecting ions of a particular m/z value and subjecting the selected
ions to fragmentation within the mass spectrometer. Generally, these
experiments are performed successfully on two types of instrument;
those where analyzers are in series (tandem in space) such as the tri-
ple quadrupole and hybrid quadrupole-TOF configurations described
earlier in this section; and secondly those instruments which employ
ion trapping mechanisms such as the quadrupole ion trap and FT-
ICR analyzer (tandem in time).
3.3 Generating MS Data for Protein Identification 243
Typically, fragmentation is performed by collision induced dissocia-
tion (CID), a mechanism of fragmenting molecular ions in the gas
phase. Peptide molecular ions are allowed to collide with neutral gas
molecules (helium, nitrogen or argon) within a cell in the mass spec-
trometer. As a result of the collision, some of the kinetic energy pos-
sessed by the molecular ion is converted into internal energy which
results in bond breakage and the fragmentation of the molecular ion
into smaller fragments.
Traditionally, three types of MS/MS experiments are performed
routinely within proteomics to determine peptide sequence and sites
of post-translational modifications (i.e. protein identification and
characterization). The configuration used in triple quadrupole- and
hybrid quadrupole-TOF instruments will be used to explain the three
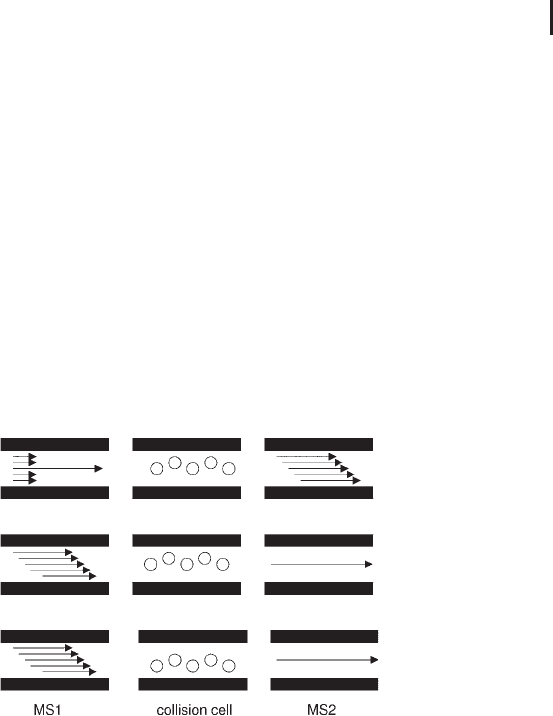
types of experiment (see Figure 3.19). Effectively the analyzer region
of these instruments can be regarded in three parts: MS1, the colli-
sion cell and MS2.
Fig. 3.19: Schematic of the three types of MS/MS experiment.
Top panel: product ion scan; middle panel: parent ion scan;
bottom panel: neutral loss scan.
3.3.4.1 Product Ion Scan
In a product ion scan, the first part of the analyzer, MS
1
, is used to
specifically select the ion of interest, the precursor ion (i.e. a peptide).
The precursor ion is allowed into the collision cell where it undergoes
collision induced dissociation (CID) (Figure 3.19, upper panel). Here,
the peptide precursor ions collide with molecules of the collision gas
yielding a distribution of fragment ions, or product ions. These pro-
duct ions are resolved by the third part of the analyzer, MS
2
, before
detection at the detector producing a product ion spectrum. With
respect to a triple quadrupole analyzer, MS
2
is a mass filtering quad-
rupole and scans the selected mass range to enable detection of the
3 Mass Spectrometry244
product ions. In the case of a hybrid quadrupole TOF analyzer, MS
2
is
a reflectron TOF analyzer, in which the entire mass range is not
scanned, but collected simultaneously with significant improvements
in sensitivity and resolution. With respect to the TOF/TOF configura-
tion, the first TOF acts as MS1 and the second TOF acts as MS
2
. Fig-
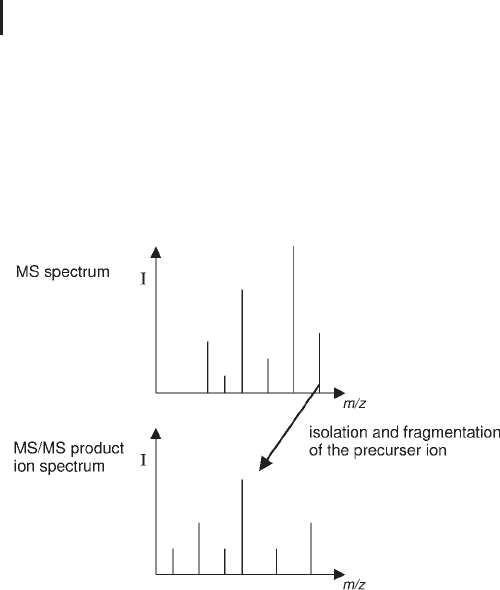
ure 3.20 demonstrates the experimental workflow, where an ion of
interest is selected and isolated by MS1 and fragmented, yielding the
product ion MS/MS spectrum.
Fig. 3.20: Product ion MS/MS experimental workflow.
Collision induced dissociation (CID) can be further defined by the
energy of the collisions observed in the collision cell. Fragmentation
within triple quadrupole, quadrupole ion trap and hybrid quadrupole
TOF analyzers occurs at collisional energies in the order of 10–100 eV
range, whilst fragmentation within a magnetic sector or TOF/TOF
analyzer occurs at collisional energies at least an order of magnitude
higher in the keV range. The former is described as low-energy CID
whilst the latter is described as high-energy CID.
As a result of the low-energy collisional fragmentation, the peptide
precursor ion fragments predictably at each peptide amide bond
along the peptide backbone yielding a distribution of product ions in
two complementary ion series forming a ladder which is indicative of
the peptide sequence. This fragmentation nomenclature was
described by Roepstorff and Fohlman (1984; see Figure 3.21). The
two complementary ion series are:
.
N-terminal ion series, or b-ion series. The ions
of the n-terminal ion series will contain the
N-terminal amino acid and extensions from this
residue (Figure 3.22 and Table 3.2).
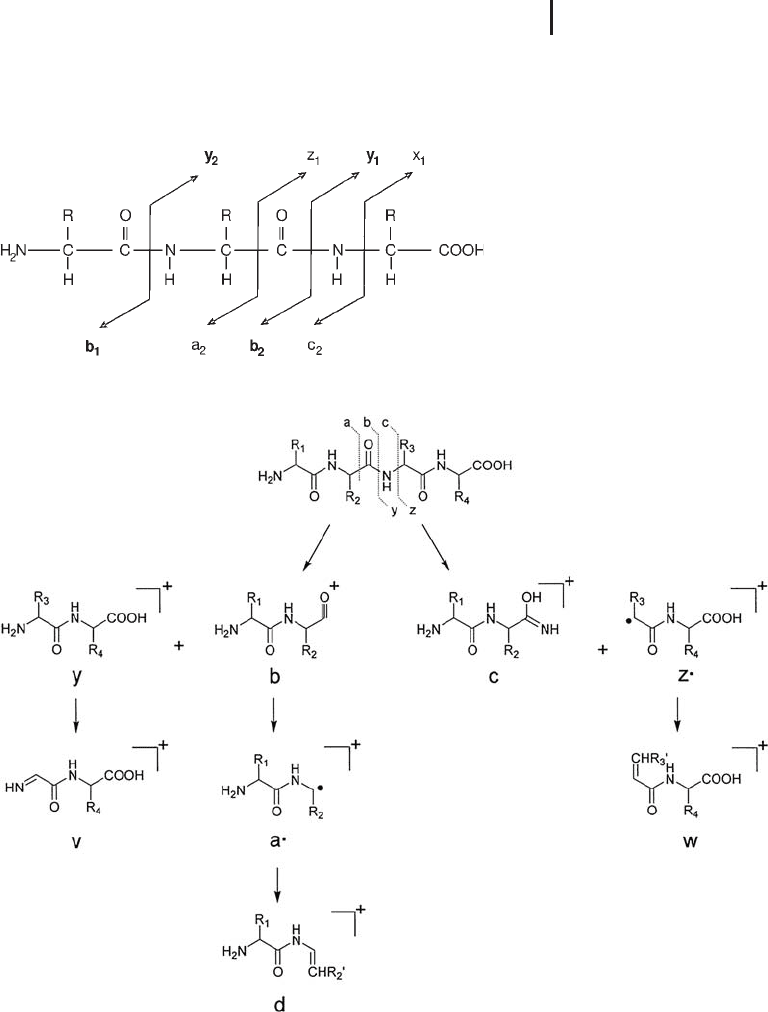
3.3 Generating MS Data for Protein Identification 245
.
C-terminal ion series, or y-ion series. The ions of
the C-terminal ion series will contain the C-ter-
minus of the peptide and extensions from this
residue (Figure 3.21 and Table 3.2).
Fig. 3.21: Fragmentation nomenclature acc. to Roepstorff and Fohlmann.
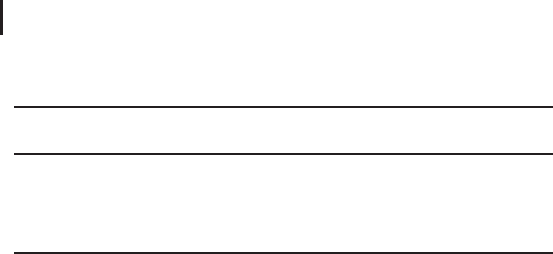
Fig. 3.22: Structure of the observed b, y, a., c, and z. ions and
d, w, and v side-chain fragment ions. R2¢ and R3¢ represent partial
side-chain loss (from. Cournoyer et al. 2005).
Roepstorff P, Fohlman J.
Biomed Mass Spectrom 11
(1984) 601.
Cournoyer JJPittman JL, Ivleva
VB, Fallows E, Waskell L, Cost-
ello CE, O'Connor PB. Protein
Sci. 14 (2005) 452–463.
3 Mass Spectrometry246
Tab. 3.2: Common fragment ions formed during low-energy CID and
the respective formulae to calculate the mass of each ion.
Fragment ion Formula
a-ion Total residue mass of the amino acids present –28
(for the loss of CO)
b-ion Total residue mass of the amino acids present
y-ion Total residue mass +18 +1
At higher collisional energies, the peptide is additionally fragmen-
ted at the amino acid side-chains (Johnson et al. 1988), the subse-
quent fragmentation pattern can be used to differentiate the isobaric
reisudes, leucine and isoleucine. Similarly, electron capture dissocia-
tion, an alternative fragmentation technique, delivers similar side-
chain specific fragmentation. (see Section 3.3.4.7).
With the knowledge of how peptides fragment within a mass spec-
trometer, the subsequent sequence information derived from product
ion MS/MS (or PSD experiments) can be used in three ways:
.
Manual interpretation of the sequence (generally
from the b- and y-ion series of fragment ions)
where the determined sequence can be inputted
as a search query (Perkins et al. 1999).
.
Manual interpretation of a short stretch of
sequence used together with the accurate mass
of the precursor ion and the masses of the first
and last fragment ions in the stretch of the
sequence (knowledge of these two masses, gives
the masses of the uninterpreted sequence in the
peptide), a method termed the peptide sequence
tag (Mann and Wilm 1994).
.
The automatic, uninterpreted MS/MS search
(Eng et al. 1994; Yates et al. 1995; Perkins et al.
1999). The process of selecting, isolating and
fragmenting ions of interest is now routinely
performed in an automated fashion, following
earlier publications demonstrating the ability to
perform data dependent fragmentation (Spahr
et al. 2000).
Johnson RS, Martin SA,
Biemann K. Int J Mass Spec-
trom Ion Proc 86 (1988)
137–154.
Perkins DN, Pappin DJC,
Creasy DM, Cottrell JS. Electro-
phoresis 20 (1999) 3551–3567.
Mann M, Wilm M. Anal Chem
66 (1994) 4390–4399.
Eng JK, McCormack AL, Yates
JR III. J Am Soc Mass Spec-
trom. 5 (1994) 976–989; Yates
JR III, Eng JK, McCormack AL,
Schieltz D. Anal Chem 67
(1995) 1426–1436.
Spahr CS, Susin SA, Bures EJ,
Robinson JH, Davis MT,
McGinley MD, Kroemer G,
Patterson SD. Electrophoresis
21 (2000) 1635–1650.

3.3 Generating MS Data for Protein Identification 247
As sequence information is significantly more discriminating than a
molecular mass alone, a few peptides of sufficient length are suffi-
cient to identify a protein from a well characterized genome (Susin
et al. 1999; Shevchenko et al. 2000).
Using product ion MS/MS as a protein ID tool, less peptides are
generally required to unambiguously identify a protein from a known
genome than with MALDI PMF. Resultingly, today’s proteomics
research uses this particular MS/MS technique in almost all protein
identification strategies which generate peptide sequence informa-
tion.
However, even a product ion MS/MS analysis may not generate
unambiguous protein identification from a protein database search
query. In these cases, a further tool for protein identification is the
expressed-sequence tag (EST) databases (Mann, 1996). ESTs are short
stretches of nucleotide sequence, which peptide sequence data can be
used to search against. Peptide sequence data can be used to search
the EST databases if a search against the protein database is unsuc-
cessful. A PMF search against the EST database is inappropriate, as it
would not be sufficiently discriminating for unambiguous identifica-
tion.
In the rarer instances, whereby a protein is not identified from any
database search query then genuine de novo sequencing needs to be
performed.
De novo sequencing requires the analysis of a peptide fragmenta-
tion spectrum without recourse to a protein database. Other applica-
tions for de novo sequencing include the characterization of splice var-
iants. Though the peptide sequences probably exist in the database,
they may not exist in a consecutive order. Thus the sequencing of
peptides, which span two exons, could be very useful for determining
splice variants (B. Kster, personal communication).
However, this is not a straightforward task. The fragmentation pat-
tern of a peptide in a product ion MS/MS spectrum is indicative of its
sequence. If a significant number of continuous residues can be
determined from the spectrum (greater than 12 residues) oligonu-
cleotide probes can be developed. However, under low-energy CID
conditions the peptide precursor ion fragments to yield y- and b-ion
series of varying lengths. Neither series may be long enough to gener-
ate the sufficient stretch of continuous residues required for subse-
quent oligonucleotide probe construction. In such circumstances it is
important to detect both ion series in the spectrum, as they are com-
plementary. For instance, the residue indicated by the first b-ion, the
N-terminal residue, b
1
, is the same residue indicated by the final y-
ion and vice versa. In this manner, the complementary ion series can
be used to fill in the gaps that may exist in the sequence of one of the
ion series.
Once a CID spectrum has been
matched, a theoretical tryptic
digest of the identified protein
can be generated and the
presence of other tryptic
peptides can be checked and
confirmed.
Susin SA, Lorenzo HK,
Zamzami N, Marzo I, Brothers
G, Snow B, Jacotot E, Constan-
tini P, Larochette N, Goodlett
DR, Aebersold R., Pietu G,
Prevost MC, Siderovski D,
Penninger J, Kroemer G.
Nature 397 (1999) 441–446.
Shevchenko A, Loboda A,
Shevchenko A, Ens W,
Standing KG. Anal Chem 72
(2000) 2132–2141.
Mann M. Trends Biol Sci. 21
(1996) 494–495.
This is often the case if the
genome of the organism in
question is not fully sequenced
or poorly characterized.

3 Mass Spectrometry248
However, the presence of two ion series can complicate the inter-
pretation of the spectrum, even though they are complementary.
Thus, for efficient de novo sequencing it is very important, if not
essential, to be able to differentiate these two series. Once this has
been achieved the presence of the two ion series is not a burden, the
interpretation of the spectrum is simplified significantly.
A range of different approaches have been taken to perform de novo
sequencing by mass spectrometry. The first report of peptide de novo
sequencing by ESI mass spectrometry was described by Hunt and
colleagues as early as 1986, where tryptic peptides of apolipoprotein B
were successfully sequenced using FAB-triple quadrupole mass spec-
trometry (Hunt et al. 1986). In this method, the group used differen-
tial modification of the peptides to differentiate each ion series, sim-
plifying interpretation and confirming the peptide sequence. Specifi-
cally, product ion MS/MS spectra were acquired of the native pep-
tides. Subsequently the peptide fractions underwent methyl esterifi-
cation, and the product ion MS/MS spectra of the corresponding deri-
vatized peptides were acquired. This reaction esterified the C-termi-
nus of each peptide (and all subsequent acidic resides in each pep-
tide, aspartic and glutamic acids). Resultantly, the starting point of
the y-ion series (C-terminal containing ions), y
1
, was shifted by 14 Da
(and hence the remainder of the y-ion series) whilst the starting point
of the b-ions series remained unchanged (unless the n-terminal resi-
due, b
1
-ion was a glutamic or aspartic acid). Hence, the y-ion series
became recognizable and the two ion series could be differentiated.
A second approach described the incorporation of an isotopic label
during the peptide digestion. Specifically, the protein of interest is
digested as normal but in a 1:1 mixture of
16
O:
18
O digestion buffer.
Subsequently all peptides will appear as
16
O:
18
O isotope doublets;
with the label being incorporated into the carboxyl group of the
C-terminal residue. When this isotopic doublet is fragmented during
a product ion MS/MS experiment, all C-terminal product ions, the
y-ion series, will exhibit this isotopic doublet and hence can be imme-
diately differentiated from the b-ion series and other non C-terminal
containing fragment ions.
Wilm and co-workers developed this approach further, combining
the isotopic labeling with a technique calling differential scanning
enabling improved assignment of the y-ion series (Uttenweiller et al.
2001). The method requires two product ion MS/MS spectra to be
acquired; one where the whole
16
O:
18
O peptide envelope is selected
for fragmentation and the second where only the
18
O labeled ions are
selected for fragmentation. Using a software algorithm the y-ion ser-
ies can be filtered automatically.
However, both approaches do not affect the fragmentation pattern
and hence the b-and y-ion series still fragment in the same manner.
Hunt DF, Yates JR III, Shaba-
nowitz J, Winston S, Hauer CR.
Proc Natl Acad. Sci USA 83
(1986) 6233–6237.
However, with this method the
sample has too be split and two
product ion MS/MS experi-
ments are required for each
peptide. The presence of
aspartic or glutamic residues
internal to the sequence would
also change by the esterifying
mass, and subsequent y- and
b-ions internal to the sequence
Schnolzer M, Jedrzejewski P,
Lehman WD. Electrophoresis
17 (1996) 945–953.
Qin J, Herring CJ, Zhang X.
Rapid Commun. Mass Spec-
trom 12 (1998) 209–216.
Shevchenko A, Chemushevich
IV, Ens W, Standing KG,
Thomson B, Wilm M, Mann
M. Rapid Commun Mass Spec-
trom 11 (1997) 1015–1024.
Uttenweiler-Joseph S,
Neubauer G, Christoforidis S,
Zerial M, Wilm W. Proteomics
1 (2001) 668–682.

3.3 Generating MS Data for Protein Identification 249
Furthermore, both approaches are only applicable to enzymatic meth-
ods and to proteins which can be digested; it is not applicable to those
samples that are presented, such as MHC class peptides.
A wide range of peptide derivatization protocols have been reported
for altering the peptide fragmentation pattern and fragmentation effi-
ciency in order to aid interpretation of product ion MS/MS spectra.
Derivatization of the peptide digest at the N-terminus with phospho-
nium quaternary tags improved sensitivity and yielded a single ion
series, the b-ion series (Roth et al. 1998); Munchbach et al. 2000;
described a derivatization protocol to aid the de novo sequencing of
peptides.
Keough and co-workers developed a strategy to obtain a single y-ion
series, particularly for PSD analysis, with impressive results (see Sec-
tion 3.3.4.4). Long stretches of consecutive y-ions from the y-ion ser-
ies can be readily observed with this technique and has the potential
to be useful for de novo sequencing. However, as only one of the ion
series is observed a long uninterrupted sequence is necessary.
Another derivatization approach to facilitate de novo sequence ana-
lysis is the derivatization of peptide digests with basic NHS esters.
Pyridyl quaternary NHS ester reagents were reported for the derivati-
zation of peptide digests for PSD analysis facilitating charge localiza-
tion at the N-terminus, subsequently yielding only the b-ion series.
(Spengler et al. 1997; Cardenas et al. 1997). These NHS ester reagents
were further developed to produce a series of gas phase basic reagents
to quantitatively label the peptide digest (N-succinimidyl morpholino
acetate and N-succinimidyl pyridyl acetate; SMA and SPA respec-
tively). Each of these tags defined the starting points for both the b-
and y-ion series, enabled the observation of often complete y-ion ser-
ies for tryptic peptides (the final y-ion is observed in all cases) and
long stretches of the b-ion series. Hence, both the starting and end
points of the peptide are known and the ion series are subtley differ-
entiated. Armed with these pieces of information de novo sequencing
is significantly simplified. Furthermore, because the two ion series
are complementary the determined sequence can be readily checked
in each ion series. This proved highly beneficial for uninterpreted,
automated MS/MS database searching and essential for de novo
sequencing (Tugal et al. 1998; Kondo et al. 1997; Hoess et al. 1999).
In today’s research, MS suppliers provide automated de novo
sequencing software to support this application. Automated de novo
sequencing is supported considerably by high mass accuracy and
high resolution improves considerably the results, providing higher
confidence in assignment of the correct amino acid sequence.
Roth KDW, Huang ZH, Sada-
gopan N, Throck Watson J.
Mass Spectrom Reviews 17
(1998) 255–274.
Munchbach M, Quadroni M,
James P. Anal Chem 72 (2000)
4047–405.
Spengler B, Luetzenkirchen F,
Metzger S, Chaurand P, Kauf-
mann R, Jeffrey W, Bartlet-
Jones M, Pappin DJC. Int J
Mass Spectrom Ion Proc 169
(1997) 127–140.
Cardenas MS, Van der Heeft E,
De Jong APJM. Rapid commun
Mass Spectrom. 11 (1997)
1271–1278.
Tugal T, Zou-Yang XH, Gavin
K, Pappin D, Canas B,
Kobayashi R, Hunt T, Stillman
B. J Biol Chem 273 (1998)
32421–32429.
Kondo H, Rabouille C,
Newman R, Levine TP, Pappin
D, Freemont P, Warren G.
Nature 388 (1997) 75–78.
Hoess M, Robins P, Naven TJP,
Pappin DJC, Sgouros T, Lindahl
T. EMBO J 18 (1999) 3868–
3875.