Westermeier R., Naven T., H?pker H.-R. Proteomics in Practice: A Guide to Successful Experimental Design
Подождите немного. Документ загружается.


3 Mass Spectrometry250
3.3.4.2 Precursor Ion Scan
For proteomics applications, a precursor ion scan is employed to
locate sites of protein modifications, such as phosphorylation. In this
experiment the first part of the MS analyzer, MS
1
, is set to transmit
all the components of the mixture in to the collision cell to undergo
CID. The third part of the analyzer, MS
2
, is fixed at a specific mass
value, so that only analytes, which fragment to give a fragment ion of
this specific mass will be detected at the detector (see Figure 3.19,
middle panel). In this manner, the precursor ion scan can be used to
selectively identify certain species in a complex mixture, such as pep-
tides containing a phosphorylated residue.
The presence of serine, threonine or tyrosine phosphorylated pep-
tide in a complex peptide digest can be determined using a precursor
ion scan. A phosphorylated peptide fragments during low-energy col-
lisional fragmentation to give a fragment ion of 79 Da (the PO
3
–
group). Therefore to specifically identify phosphorylated peptides in a
mixture the third part (MS
2
) of the analyzer is set to 79. Thus, only
species which fragment to give a fragment ion of 79 are detected and
hence indicate the presence of a phosphorylated residue (Wilm et al.
1996; Carr et al. 1996; Annan et al. 2001).
This experiment has been historically restricted to a triple quadru-
pole instrument, but recent applications have been demonstrated
using a hybrid quadrupole TOF instruments (Steen et al. 2001). The
method has been exploited for the specific detection of tyrosine phos-
phorylated peptides in complex mixtures.
3.3.4.3 Neutral Loss Scan
Similarly, a neutral loss scan is employed in proteomics applications
to locate peptides containing a protein modification. In this techni-
que, the 1
st
and 3
rd
parts of the analyzer are scanned synchronously,
but with a specific m/z offset. Once more the second part of the ana-
lyzer is used as the collision cell and the entire mixture is allowed to
enter the collision cell, only those species which fragment to yield a
fragment ion with the same mass as the offset will be detected (see
Figure 3.20). For instance, serine and threonine phosphorylated pep-
tides readily lose phosphoric acid during low-energy CID. Phosphoric
acid has a mass of 98 Da, thus the offset for a doubly charged peptide
would be set at 49. Hence any species which loses 49 Da from a dou-
bly charged ion would be observed at the detector and be indicative of
a phosphorylated peptide (Covey et al. 1991; Schlosser et al. 2001).
This type of experiment is typically carried out with a triple quad or
Q-TOF type instrument. Phosphopeptide analysis in an ion trap is
Wilm M, Neubauer G, Mann
M. Anal Chem. 68 (1996)
527–533.
Carr SA, Huddleston MJ,
Annan RS. Anal Biochem 239
(1996) 180–192.
Annan RS, Huddleston MJ,
Verna R, Deshaies RJ, Carr SA.
Anal Chem 73 (2001) 393–
404.
Steen H, Kster B, M
Fernandez, Pandy A, Mann M.
Proceedings of the 49
th
ASMS
conference on mass spectro-
metry and allied topics,
Chicago ( 2001).
Covey TR, Huang EC, Henion
JD. Anal Chem 63 (1991)
1193–1200.
Schlosser A, Pipkorn R, Bosse-
meyer D, Lehman WD. Anal
Chem 73 (2001) 170–176.

3.3 Generating MS Data for Protein Identification 251
typically performed with a MS
3
approach. Both approaches have dif-
ferent specific benefits, which are not pointed out here.
3.3.4.4 MALDI-TOF Post Source Decay
Though not a genuine MS/MS technique, MALDI-TOF post source
decay (PSD) is capable of generating sequence specific information,
which can be used for protein identification. Described by Spengler
and co-workers in the 1990s (Spengler 1997; Chaurand et al. 1999),
this technique demonstrated some reasonable success as a protein
identification tool. The quality of data is less than that acquired with
conventional MS/MS instruments, as described earlier (Silles et al.
2000; Gevaert et al. 2001), though still sufficient for protein ID from
relatively simple samples such as those derived from a 2D PAGE gel.
In the technique, the peptide of interest is specifically selected
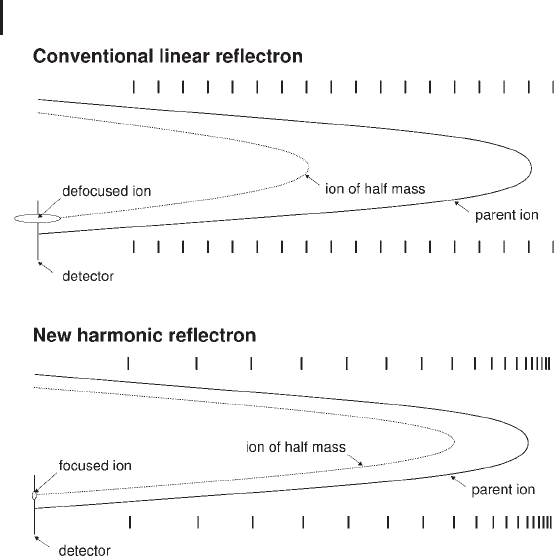
from a complex mixture using an ion gate. In a conventional linear
reflectron, the configuration can only accommodate a small range of
KE differences i.e the reflectron will only be able to focus those ions
that are similar in mass to the parent ion (Figure 3.23). In this
instance, only PSD ions that are close in mass to the precursor ion
will be mass measured correctly. Thus, to perform PSD with a con-
ventional reflectron the voltage on the reflectron has to drop sequen-
tially to allow the fragment ions of lower mass (and lower KE) to
become focussed at the detector. This is termed stepping the reflec-
tron. In a conventional reflectron, several different voltages or seg-
ments, (as many as 12 segments may be required) are required to
focus the entire mass range of product ions (Figure 3.24a). The indi-
vidual segments are then "stitched" together using the instrument
software, generating the complete product ion spectrum. (Figure
3.24b).
The use of a quadratic field reflectron for PSD analysis attempts to
address the issue of stepping the reflectron. It is capable of accommo-
dating ions of widely different kinetic energies in a single spectrum
(Figures 3.22–3.23), because in a quadratic field reflectron an
increased voltage is applied, non-linearly, to create a perfect quadratic
field across the reflectron. Resultantly, all the product ions created
during the post source decay of a particular precursor ion are focused
at the detector, irrespective of their energy, over the entire range of
m/z. Hence, a complete PSD spectrum is obtained under the same
experimental conditions with each pulse of the laser, without the
need for data stitching.
Spengler B. J Mass Spectrom.
32 (1997) 1019–1036.
Chaurand P, Luetzenkirchen F,
Spengler B. J Am Soc Mass
Spectrom 10 (1999) 91–103.
Silles E, Mazon MJ, Gevaert K,
Goethals M, Vandekerckhove,
Lebr R, Sandoval IV. J Biol
Chem. 275 (2000) 34054–
34059.
Gevaert K, Demol H, Martens
L, Hoorelbeke B, Puype M,
Goethals M, Van Damme J, De
Boeck S, Vandekerckhove J.
Electrophoresis 22 (2001)
1645–1651.
3 Mass Spectrometry252
Fig. 3.23: Schematic demonstrating the passage of the precursor ion and
product ion through two types of reflectron. Upper panel: conventional linear
reflectron. Lower panel: quadratic field reflectron.
Under typical timed ion delay MALDI conditions, peptide fragmenta-
tion observed during PSD is reduced (Kauffman et al. 1996). Keough
and co workers described a derivatization method for peptide
sequence analysis by MALDI PSD that facilitated fragmentation dur-
ing PSD, increasing sensitivity significantly. The tag was designed to
promote efficient charge-site initiated fragmentation of the peptide
bonds. Consequently, only a single ion series, the y-ion series, is
observed in the PSD spectrum, simplifying interpretation.
The theory proposed that under MALDI ionization conditions, the
strong basic residue at the C-terminus of tryptic peptides (arginine)
would be protonated and the strong acidic group at the C-terminus
would be deprotonated (Figure 3.25). The additional proton (almost
exclusively MALDI ions in the peptide mass range are singly charged)
would then be free to randomly ionize and subsequently fragment
the peptide amide bonds of the peptide backbone. Hence, when the
fragment ions are formed only the y-ions are observed because, the
formal positive charge typical of a b-ion is neutralized by the negative
charge on the sulfonic acid group, hence b-ions are not observed.
Kauffman R, Chaurand P,
Kirsch D, Spengler B. Rapid
Commun Mass Spectrom 10
(1996) 1199–1208.
Keough T, Youngquist RS,
Lacey MP. Proc Natl Acad Sci
USA 96 (1999) 7131–7136.
Keough T, Lacey MP, Fieno
AM, Grant RA, Sun Y, Bauer
MD, Begley KB. Electrophoresis
21 (2000) 2252–2265.

3.3 Generating MS Data for Protein Identification 253
Fig. 3.24: Schematic of the time focusing of a conventional and
quadratic field reflectron.
Upper panel: only a small segment of the mass range can be
focussed at one time with a conventional linear reflectron.
Middle panel: multiple segments acquired to obtain the whole
mass range of fragment ions using a conventional linear
reflectron.
Lower panel: quadratic field reflectron allows the focussing of
the mass range in a single TOF spectrum.
–
O
3
S–A–A–A–A–A–R
+
(a)
–
O
3
S–CHR–CO
+
[H
2
NCHR–CO
2
H]
+
(b)
b
1
ion y
1
ion
(R = guanidino side-chain of arginine)
Fig. 3.25: The effect of a strongly acidic group at the N-terminus
of an arginine C-terminating peptide.
3 Mass Spectrometry254
In the original method, the peptide digest was derivatized with 2-sul-
fonyl acetyl chloride, labeling the a-amino group at the N-terminus of
each peptide (and the e-amino group of the each lysine side-chain)
with the very strong acid, sulfonic acid (pKa<2).
Labeling of arginine-terminating tryptic peptides greatly facilitates
fragmentation, particularly fragmentation by MALDI PSD. Figure
3.26 demonstrates the extent of fragmentation during a PSD experi-
ment. In the case of this synthetic peptide, 16 consecutive residues
can be readily determined. The derivatization technique is compatible
with protein digests derived from 2D gels (see Sections 3.4.3–3.4.4).
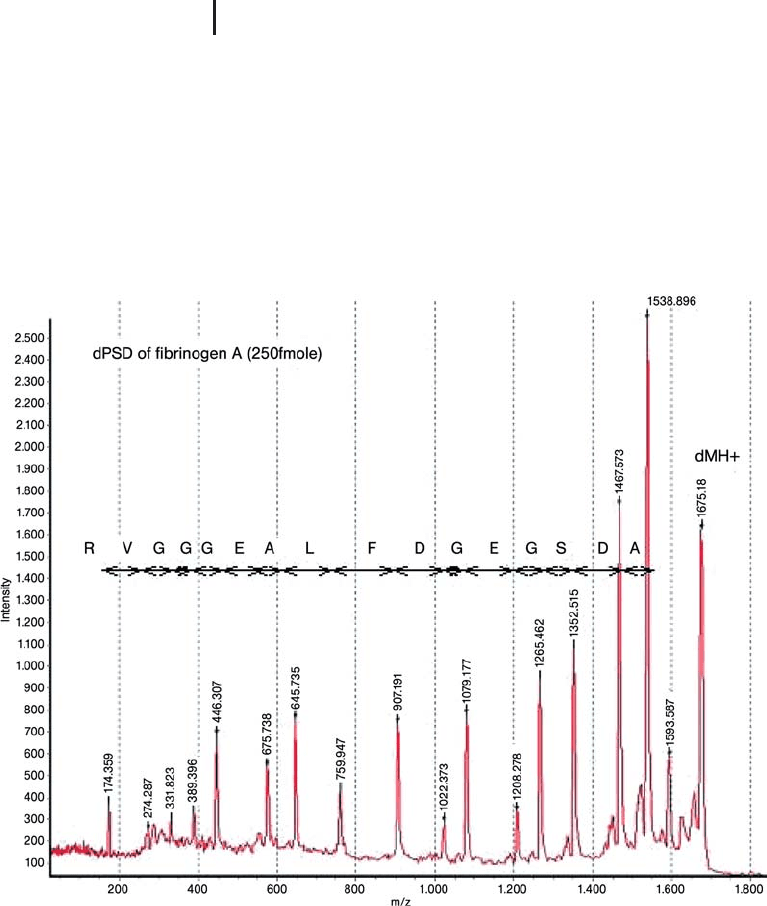
Fig. 3.26: PSD spectrum of a CAF derivatized synthetic peptide
acquired with the Ettan MALDI-TOF Pro (250 fmol applied to the target).
The 16 consecutive residues are readily determined. Though Leucine (L)
is specified in the sequence, this cannot be determined from the
experiment (Isoleucine and leucine are isobaric). dMH
+
is the precursor
ion. Note the intensity of the precursor ion. In a typical PSD experiment,
fragment ion abundance is a very low percentage of the precursor ion
abundance.
3.3 Generating MS Data for Protein Identification 255
As the derivatization strategy also causes the derivatization of the
e-amino group of the lysine side-chain, therefore labeling the lysine
with a strong acidic group, PSD of lysine C-terminating peptides
becomes inefficient. To extend the applicability of this method to
lysine terminating peptides, the lysine must first be modified to the
homo-arginine derivative. This has recently been reported by a num-
ber of groups to increase lysine tryptic peptide detection in peptide
mass fingerprinting (Brancia et al. 2000; Keough et al. 2000). If this
modification of the lysine peptides is performed prior to the sulfona-
tion reaction then the sulfonation methodology is equally applicable
to lysine C-terminating peptides.
The sulfonation method has further been improved with the devel-
opment of a water stable sulfonic NHS ester reagent (Figure 3.27;
Liminga et al. 2001). Subsequent PSD spectra acquired from peptides
derivatized in this manner is termed chemically assisted fragmenta-
tion (CAF).
N
O
O
O
O
R
S
O
O
OH
NH
2
Pep
S
O
O
OH
R
O
N
H
Pep
+
Fig. 3.27: Reaction scheme for the water stable sulfonic NHS ester reagent.
One of the drawbacks of the original method was the hygroscopic nat-
ure of the acetyl chloride reagent, requiring the derivatization proce-
dure to be performed under stringent conditions in a non-aqueous
environment. The novel watersoluble reagent allows the quantitative
derivatization of the peptide digest at sub pmol quantities of starting
material and is compatible with the lysine modification step affording
a one pot derivatization (see Step 10). The derivatization step is amen-
able to automation. This derivatization approach enables peptide
mass fingerprinting data and subsequent sequence information to be
acquired on the same instrument with good sensitivity (see Section
3.5.1).
This derivatization strategy is equally applicable to any MS/MS
instrument, which employs MALDI as the ionization source,
enabling the simple interpretation of the peptide sequence from the
fragmentation of a singly charged ion.
Despite the efforts to improve the performance of MALDI PSD, the
development of MALDI TOF/TOF systems and the performance and
automation of LC-ESI-MS/MS systems, the use of MALDI PSD has
become considerably reduced.
Brancia FL, Oliver SG, Gaskell
SJ. Rapid Commun Mass Spec-
trom 14 (2000) 2070–2073.
Keough T, Lacey MP, Young-
quist RS. Rapid Commun Mass
Spectrom 14 (2000) 2348–
2356.
Liminga M, Born M, strçm
J, Carlsson U, Keough T,
Maloisel JL, Palmgren R,
Youngquist S. Proceedings
ASMS conference on mass
spectrometry and allied topics,
Chicago, USA (2001).
3 Mass Spectrometry256
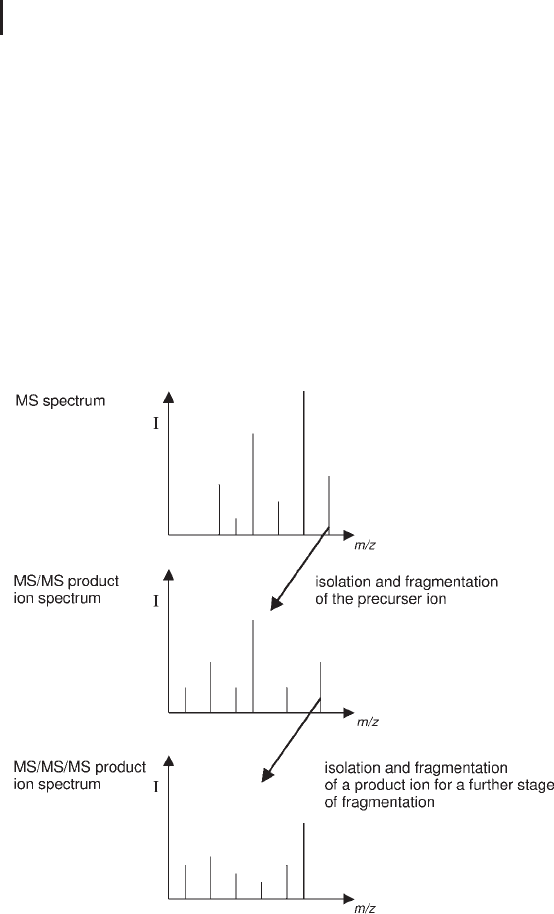
3.3.4.5 Multi-stage Tandem Mass Spectrometry
Multi-stage tandem mass spectrometry (MS
n
) involves multiple
stages of mass selection, separated by some form of fragmentation.
This type of fragmentation experiment is restricted to quadrupole ion
traps and FT-ICR MS. A fragment ion produced in the product ion
MS/MS spectrum can be selected, isolated and fragmented a second
time generating even further sequence information in a third spec-
trum. This spectrum is called an MS/MS/MS product ion spectrum,
or MS
3
(see Figure 3.28; Louris et al. 1989). This type of experiment is
useful for identification of phosphorylated or glycosylated residues if
sufficient material exists. This process can be repeated a number of
times, resulting in a series of MS
n
spectra where n’ represents the
number of times the fragmentation cycle has been performed.
Fig. 3.28: Schematic demonstrating the workflow in an MS
3
experiment.
3.3.4.6 Electron Transfer Dissociation
Electron transfer dissociation (ETD) is a new fragmentation techni-
que that fragments peptides by transferring electrons to positively
charged peptides, yielding a strech of sequence ions derived from
Louris JN, Amy JW, Ridley TY
and Cooks RG. Int J Mass
Spectrom Ion Proc 88 (1989)
97–111

3.3 Generating MS Data for Protein Identification 257
cleavage at the amide groups along the peptide backbone. Due to its
non-ergodic nature, amino acid side-chains and important modifica-
tions such as phosphorylation are not fragmented and therefore
important discriminatory information is maintained. The technique
also has the potential to analyze large peptides and proteins and
therefore support the top dpwn approach to proteomics. ETD is now
available in two commercially available ion traps.
3.3.4.7 Electron Capture Dissociation
Electron capture dissociation (ECD) is an alternative fragmentation
method to CID, but it is restricted to FT-ICR instruments (Zubarev
et al. 1998; McLafferty et al. 2000). In the ECD method, multiply
charged ions are irradiated with low-energy electrons produced by an
emitter cathode located behind the ICR cell. Upon capture of elec-
trons, reduced radical cations [M+nH]
(n–1)+
are generated which dis-
sociate by fast and facile fragmentation of the N–C
a
bond of the pep-
tide chain, producing mainly c and z fragment ions. The ECD
mechanism exhibits extensive peptide cleavage, retention of sites of
modification (Shia et al. 2001) and side-chain specific cleavage
enabling the differentiation of isobaric residues such as leucine and
isoleucine (Kjeldsen et al. 2003) by producing predominantly c and z
type fragment ions (Cooper et al. 2005); again it is non-ergodic in nat-
ure and in that sense similar to ETD.
The technique supports the top down approach to proteomics (Sec-
tion 3.6.2), where intact protein molecular ions are fragmented
within the mass spectrometer enabling protein sequence and PTM
information to be acquired without performing proteolysis.
The top-down proteomics approach is further supported by the
infra red multiphoton dissociation (IRMPD) dissociation technique
(Breuker et al. 2002). This technique is used for ion activation prior to
ECD. Ion activation prior to ECD in this fashion, results in greater
fragmentation of the protein backbone enabling more sequence infor-
mation to be determined.
Generation of the product ion MS/MS spectra alone does not iden-
tify the protein, this requires a database search query using one of the
common search engines. MS experiments for protein identification
rely heavily on computational data analysis and database searching
algorithms. These algorithms have been developed to assign peptide
sequences based on automated interpretation of MS/MS spectra (Eng
et al. 1994; Perkins et al. 1994; Fenyo et al. 2002; Creasy et al. 2002).
Zubarev RA, Kelleher NL,
McLafferty FW. J Am Chem
Soc 120 (1998) 3265–3266.
McLafferty FW, Horn DM,
Breuker K, Ge Y, Lewis MA,
Cerda B, Zubarev RA,
Carpenter BK. J Am Soc Mass
Spectrom. 12 (2001) 245–249.
Shi SDH, Hemling ME, Carr
SA, Horn DM, Lindh I, McLaff-
erty FW. Anal Chem. 73
(2001) 19–22.
Kjeldsen F, Haselmann K F,
Sorensen ES, Zubarev RA..
Anal. Chem. 75 (2003) 1267–
1274.
Cooper HJ, Hkansson K,
Marshall AG. Mass Spectrum.
Reviews 24 (2005) 201–222.
Breuker K, Oh H, Horn DM,
Cerda B, McLafferty, FW. J Am
Chem Soc 124 (2002) 6407–
6420.
Eng JK, McCormack AL and
Yates JR 3
rd
, J Am Soc Mass
Spectrom 5 (1994) 976–989.
Perkins DN, Pappin DJC,
Creasy DM, Cottrell JS. Electro-
phoresis 20 (1999) 3551–3567.
Fenyo D, Beavis RC.Trends
Biotechnol. 20 (2002) 35–38.
Creasy DM, Cottrell JS. Proteo-
mics 2 (2002) 1426–1434.

3 Mass Spectrometry258
Automated analysis of peptide MS/MS spectra is now routine with
hundreds of MS/MS spectra being analyzed in a single LC-MS run.
However, with hundreds of assignments being returned by the search
query for every LC-MS run it is important to be confident that the
assignment is correct and not a false identification. For a comprehen-
sive review of database mining for proteomics applications, please
refer to the monography of Lisacek.
3.4
Protein Characterization
Protein characterization extends further than simply protein identifi-
cation and often refers to the identification and location of post-trans-
lational modifications (PTMs). PTM analysis is one of the pillars on
which proteomics is built, as PTM information cannot be determined
at the DNA level. Post-translational modification of proteins is an
essential route to modify the function/activity of that particular pro-
tein, with the process being governed by a series of enzymes includ-
ing kinases and phophatases. There are several hundred examples of
post-translational modifications (Garavelli, 2004), but the most com-
mon modifications studied are reversible phosphorylation and glyco-
sylation Specifically, reversible phosphorylation of proteins is a key
mechanism for the regulation of major cellular processes such as pro-
liferation, differentiation or apoptosis through highly dynamic and
complex signalling pathways. It has been estimated that 100,000
potential phosphorylation sites exist in the human proteome, with
the large majority poorly characterized (Zhang et al. 2002).
The key challenges in PTM analysis are to isolate/enrich for the
modified protein/peptide from a complex mixture, to quantify the
phospoprotein expression, determine the stoichiometry or occupancy
and to pinpoint the site of modification. To further complicate the
analysis, post-translationally modified proteins are often expressed
with low abundance. Traditionally, PTMs have been identified using
radiolabeling and Edman sequence analysis, amino acid analysis and
immunochemistry. However, due to the low protein expression of tar-
get proteins and the complexity of starting samples methods with
greater sensitivity of detection for PTMs are required.
As we have described previously in this chapter, the evolution of
mass spectrometry instrumentation over the last 20 years has had a
sigificant impact on protein ID and proteomics. Moreover, mass spec-
trometry is well placed to support the identification of PTMs.
Lisacek, F. A guide of good
practices for protein-centric
bioinformatics. WILEY-VCH,
Weinheim (2007).
Garavelli JS. Proteomics 4
(2004) 1527–1533.
Zhang H, Zha X, Tan Y, Horn-
beck PV, Mastrangelo AJ, Alessi
DR, Polakiewicz RD, Comb
MJ. J Biol Chem 277 (2002)
39379–39387.

3.4 Protein Characterization 259
& MS provides the sensitivity needed to study PTMs,
though the sample preparation prior to MS is
absolutely essential to benefit from this sensitivity
capability.
As a post-translational modification will cause a change in the mass
of protein, specifically to the amino acid it is attached to, mass spec-
trometry can detect this. Further, tandem mass spectrometry techni-
ques such as a precursor ion scan and a neutral loss scan can be used
to identify peptides that contain a certain PTM within a complex mix-
ture. Once the PTM containing peptide has been detected, a product
ion tandem mass spectrometry scan can be performed to determine
the location of the PTM within that particular peptide (see Fig. 3.19).
3.4.1
Phosphorylation Analysis
Protein phosphorylation is arguably the most studied PTM. Unlike
N-linked glycosylation, there is no specific sequon which indicates
phosphorylation, though phosphorylation commonly occurs on ser-
ine, threonine and tyrosine and less commonly on histidine residues.
Many methods have been reported and examples exist where the state
of phosphorylation of a single protein has been determined. However,
the mapping of an entire phosphoproteome and relative quantitative
analysis of phosphoproteins could be considered something of a holy
grail in current proteomics method development and here has been
considerable development in pursuit of this goal (Sachon et al. 2006;
Goshe et al. 2001; Blagoev et al. 2004; Gruhler et al. 2004; Tao et al.
2005; Zhou et al. 2001; Reinders et al. 2005).
Strategies that are specifically designed to identify sites of phos-
phorylation, attempt to isolate or at least enrich for phosphopeptides
or phosphoproteins upstream of tandem mass spectrometry.
& Tandem mass spectrometry is the key tool for
identifying the site of phosphorylation.
The isolation/enrichment method is either based on an affinity chro-
matography step or a chemical derivatization approach and most
approaches to date are applied at peptide level (Figure 3.29).
Sachon E, Mohammed S,
Bache N, Jensen ON. Rapid-
Commun Mass Spectrom 20
(2006) 1127–1134.
Goshe MB, Conrads TP,
Panisko EA, Angell NH, Veen-
stra TD, Smith RD. Anal Chem
73 (2001) 2578–2586.
Blagoev B, Ong SE, Kratch-
marova I, Mann M. Nat
Biotechnol 22 (2004) 1139–
1145.
Gruhler A, Olsen JV,
Mohammed S, Mortensen P,
Færgeman NJ, Mann M,
Jensen ON. Mol Cell Proteo-
mics 4 (2005) 310–327.
Tao WA, Wollscheid B, O’Brien
R, Eng JK, Xiao-jun L, Boden-
miller B, Watts JD, Hood L,
Aebersold R Quantitative phos-
phoproteome analysis using a
dendrimer conjugation chem-
istry and tandem mass spectro-
metry. Nat Methods 2 (2005)
591–598.
Zhou H, Watts JD, Aebersold
R. Nat Biotechnol. 19 (2001)
375–337.
Reinders J, Sickmann A.
Proteomics 5 (2005) 4052–
4061.
Though for some simpler
samples, it maybe possible to
employ a shotgun method,
fractionating the sample prior
to tandem mass spectrometry.