Brewster H.D. Solid State Physics
Подождите немного. Документ загружается.


12 Energy Dispersion Relations in Solids
For simplicity, we will limit the present discussion
of
the tight binding
approximation to
s{bands (non degenerate
atomic states) and therefore we can suppress
the
j index on the wave functions. (The treatment for p-bands
is
similar to
what we will do here, but more complicated because
of
the degeneracy
of
the
atomic states.) To find matrix elements
of
the Hamiltonian we write
(k'IHlk)=lsI
2
:L>l(k-R
n
)
f~*(r-RIll)H~(r-Rn)d3r
n,m
Q
in
which the integration
is
carried out throughout the volume ofthe crystal.
Since
H
is
a function which
is
periodic
in
the lattice, the only significant
distance
is
(Rn
-
Rm
) = P
11111
We then write the integral
in
equation as:
(f'1
HI
k)
=1
S
12
~>i(f.f').Rm
I
i~Pnl/1
Hmn(Pnm)
Rm
P
nm
where we have written the matrix element
Hmn
(Pnm)
as
Hmn
(P
nm
) = f f
(r
-
RnJH
~(r
-
Rm
-
Pnm
d3r
= f f
(r'
-
Pnm
)d3
r
,
Q Q
We note here that the integral in equation depends only on
Pnm
and not
on
Rm
. According to equation, the first sum
in
equation IS
"ei(k-f').R
m
=0-
-
-N
L.
k',k+G
Rm
where G is a reciprocal lattice vector. It is convenient to restrict the k
vectors to lie within the first Brillouin zone (i.e., we limit ourselves to reduced
wave vectors). This is consistent with the manner
of
counting states with the
periodic boundary conditions on a crystal
of
dimension d on a side
kid
=
2TCmi
for each direction i
where m
i
is
an integer in the range I
::;
m
i
< Ni where Ni Z Nl=3. From equation
we have
2nml
k=
--.
I d
The maximum value that a particular m
i
can assume
is
Ni and the maximum
value for
k
i
is
2TC
= a at the Brillouin zone boundary since
N/d
=
11
a.
With
this restriction,
k and
k'
must both lie within the
p!
B.Z. and thus cannot
differ by any reciprocal lattice vector other than
G =
O.
We thus obtain the
following form for the matrix ment
of
H (and also for the matrix elements
Ho
and
H'):

Energy Dispersion Felalions
in
Solids
13
I
k
-, 1
HI
k-)
-I
j:
12
N8-
-
'"
if-p/1//1
H
(-
)
\
-~
k.k'L..
e
mn
P
nm
yielding the result
_
1-
-)
I _
eli:
P
m
"H
ml1
(Pn111)
E(
k)
=
\k
l}f 1 k =
_P-,=I1I',---'
-::-_----
Iklk)
'"
lk'PlImS
(-
)
\ L..
PIIIII
e
llln
P
nm
in
which
\k'i
k)
=1
S
12
8
k
.k,NI
i:
PI/III
SIl1l1(P11l1l)
PI/III
where the matrix element
Smn
(Pnlll)
measures the overlap
of
atomic
functions on different sites
Smn(PnnJ =
f~*
(r)~(r)~(r
- PnnJd
3
r.
n
The overlap integral
Smn
(Pnm)
will be nearly I when P
nm
= 0 and will
fall
off
rapidly as P
nm
increases, which exemplifies the spirit
of
the tight
binding approximation.
By
selecting k vectors to lie within the first Brillouin
zone, the orthogonality condition on the
'tj;
k
(r)
is automa-tically satisfied.
Writing
H =
Ho
+ H' yields:
or
f
* - [
112
2
-]
- 3
Hmn
=
~
(r
-~)
--V'
+U(r
-Rn)
~(r
-Rn)d
r
n 2m
f~*
(r
-
Rm)[V(F)-U(r
-Rn)]~(r
-Rn)d
3
r
n
H
mn
=
E(O)
Smn
(P
nm
) +
H~n
(Pnm)
which results
in
the general expression for the tight binding approximation:
'"
ik·p
H'
(-
)
_ (0) L..
p
-
e nm
mn
P
nm
E(k)=E
+ nm
.__
•
'"
lk·p S
(-
)
L..-
e nm
mn
P
nm
P
nm
In the spirit
of
the tight binding approximation, -the second term in
equation
is
assumed to be small, whi'ch is a good approximation
if
the overlap
ofthe
atomic wave functions is small. We classify the sum over
Pnm
according
to the distance between site
m and site n: (i) zero distance, (ii) the nearest
neighbour distance, (iii) the next nearest neighbour distance, etc.
I e
ik
,
p
l/lII
H~n(Pnm)
=
H~n(O)
+ Ieik'PIlIII
H~n(Pnm)
+ ...
P
IIIII
PI
The zeroth neighbour term
H'nn(O)
in
equation results in a constant

14
Energy Dispersion Relations in Solids
additive energy, independeilt
of
k . The sum over nearest neighbour distances
PI
gives rise to a k -dependent perturbation, and hence
is
of
particular interest
in
calculating the band structure. The terms
H'nn(O)
and the sum over the nearest
neighbour terms
in
equation are
of
comparable magnitude, as can be seen by
the following argument. In the integral
H:
lI1
(O)
=
Jf
(r
- Rn)[V -
U(r
- Rn)]$(r - Rn)d
3
r
we note that 1
$(r
-
Rn)
12
has
an
appreciable amplitude only
in
the
vicil),ity
of
the site
Rn'
But at site
Rn'
the potential energy term [V -
U(r
-
Rn)]
=
H'
is
a small term, so that
H:
1I1
(0) represents the product
of
a small term times a
large term.
On the other hand, the integral
H~l/l(Pnm)
taken over nearest
neighbour distances has a factor
[V -
U(r
- Rn)] which
is
large near the
mth
site; however, in this case the wave functions
$*
(r
-
Rn)
and
$(r
- Rn) are
on different atomic sites and have only a small overlap on nearest
neighbour'
sites. Therefore
H'mn
(P
nm
) over nearest neighbour sites also results
in
the
product
of
a large quantity times a small quantity.
In treating the denominator
in
the perturbation term
of
equation, we must
sum
I
i"Pnm
Smn
(P
nm
) =
Snn
(0) + I
i"Pnm
Smn
(Pnm)
+ ...
Pnm
PI
In this case the leading term
Snn(O)
is
approximately unity and the overlap
integral
Smn
(Pnm)
over nearest neighbour sites
is
small, and can be neglected
to lowest order in comparison with unity. The nearest neighbour term in
equation
is
of
comparable magnitude to the next nearest neighbour terms
arising from
Hmn(Pnm)
in
equation.
We will
here
make
several
explicit
evaluations
of
E
(k)
in
the
tight {binding limit to show how this method incorporates the crystal symmetry.
For illustrative purposes we will give results for the simple
cubic lattice (SC),
the body centered cubic (BCC) and face centered cubic lattice (FCC). We
shall assume here that the overlap
of
atomic potentials on neighboring sites is
sufficiently weak so that only nearest neighbour terms need be considered
in
the sum on
HOmn
and only the leading term in the sum
of
Smn'
For the simple cubic structure there are 6 terms in the nearest neighbour
sum on
H'mn
with
PI
vectors given by:
PI
= a(±l,O,O),a(O,±l,O),a(O,O,±l).
By
symmetry
H'mn
(PI) is the same for all
of
the
PI
vectors so that
E(k)
=
E(O)
+
H~n(O)
+
2H~n(PI)[coskxa
+
coskya
+ coskza] + ...

Energy Dispersion Relations
in
Solids
Fig. The relation between the atomic levels and the
broadened level in the tight binding approximation.
15
where
PI
= nearest neighbour separation and k
r
•
k
j
.•
k:;
are components
of
wave vector k
in
the first Brillouin zone.
This dispersion relation
E(k)
clearly satisfies three properties which
characterize energy eigenvalues in typical periodic structures:
1.
Periodicity
in
k space under translation by a reciprocal lattice vector
k~k+{;,
2.
E(k)
is an even function
of
k (i.e., E(k) = E
(-k))
3.
dE/dk = 0 at the Brillouin zone boundary
In the above expression for
E(k)
, the maximum value for the term
in
brackets is ± 3.
Therefore
for a
simple
cubic lattice in
the
tight
binding
approximation we obtain a bandwidth
of
12
H'nln
(PI) from nearest neighbour
interactions. Because
of
the different locations
ofthe
nearest neighbour atoms
in the case
of
the
Bee
and FCC lattices, the expression for
E(k)
will be
different for the various cubic lattices. Thus the form
of
the tight binding
approximation explicitly takes account
of
the crystal structure. These results
are summarized below.
simple cubic
E(k)
= const +
2H'l/I
n
CPl)
[cos
k.p
+ cos
kp
+ cos
k:;a]
+
...
body centered cubic
The
eight
PI
vectors for the nearest neighbour distances
in
the
Bee
structure are
(±
a/2, ± a/2, ± a/2) so that there are 8 exponential terms which
combine in pairs such as:
[
ik a ikv
a
ik_a
-ik
a ikv
a
ik_a]
exp-x-exp-'
-exp---
+
exp--x-exp-'
-exp---
2 2 2 2 2 2
to yield
(
k
a)
ik)'a ik_a
2cos
_x_
exp-'
-exp---.
2 2 2
We thus obtain for the
Bee
structure:
-
(k
a)
(k)'a)
(k
a)
E(k)
= const +
8H~m(Pl)cos
; cos 2 cos ; + ...

16
Energy Dispersion Relations in Solids
where
H:
lln
(PI)
is the matrix element
of
the peliurbation Ham ilton-ian
taken between nearest neighbour atomic orbitals.
face centered cubic
For the FCC structure there are
12
nearest neighbour distances
PI:
(o,±~,±~
J,(
±%.±%.o
l
(±%,o.±~).
so
that
the
twelve
exponential
terms combine
in
groups
of
4 to yield:
ik.a ikja ik.a -ikj.a
-ik.o
ikja
exp
-'\-exp-'
- +
exp-x-exp
---
+
exp--'\-exp
-'
-
2 2 2 2 2 2
-ik
a -ikv
a
(k
a)
(kj.a)
+exp----:j-exp-
2
-'-=4cos
; cos 2
thus resulting
in
the energy dispersion relation
E(k)
= const + 4H:
lln
(Pl)
[cos(
k~a)
+
cos(
k;a)
+
cos(
k;a
)cos(
k;a)
+
cos(
k;a
)cos(
k~a)
1 + ...
We note that
E(k)
for the
Fee
is different from that for the
se
or
Bee
structures. The tight-binding approximation has symmetry considerations built
into its fonnulation through the symmetrical arrangement
of
the atoms in the
lattice. The situation
is
quite different in the weak binding approximation where
symmetry
enters
into
the
form
of
VCr) and
determines
which
Fourier
components
Va
will be important in creating band gaps.
Weak
and
Tight
Binding
Approximations
We will
now
make
some
general statements
about
bandwidths and
forbidden band gaps which follow from either the tight binding or weak binding
approximations. With increasing energy, the bandwidth tends to increase.
On
the tight{binding picture, the higher atomic states are less closely bound to
the nucleus, and the resulting increased overlap
of
the wave functions results
in a larger value for
H~1n(Pl)
in the case
of
the higher atomic states: that is,
for silicon, which has 4 valence electrons in the
n = 3 shell, the overlap integral
H~1n(Pl)
will be smaller than for which is isoelectronic to silicon but has
instead 4 valence electrons in the
n = 4 atomic shell. On the weak{binding
picture, the same result follows, since for higher energies, the electrons are
more nearly free; therefore, there are more allowed energy ranges available,
or equivalently, the energy range
of
the forbidden states is smaller. Also
in
the weak{binding approximation the band gap
of
21
Va
1 tends to decrease as
G increases, because
of
the oscillatory character
of
e
ia
.
r
in
Energy Dispersion Relations
in
Solids
Vc
==
_1_
J
e-
iC
"V(F)d
3
,..
no
no
17
From the point
of
view
of
the tight{binding approximation, the increasing
bandwidth with increasing energy
is
also equivalent to a decrease
in
the
forbidden band gap. At the same
time, the atomic states at higher energies
become more closely
spaced, so that the increased bandwidth eventually results
in
band overlaps. When band overlaps occur, the tight-bil;ding approximation
as given above must be generalized to treat coupled
or
interacting bands using
degenerate perturbation theory.
Tight Binding
Approximation
with 2
Atoms/Unit
Cell
We present here a simple example
of
the tight binding approximation for
a simplified version
of
poly acetylene which has
two
carbon atoms (with their
appended hydrogens) per unit cell.
V(r)
r
Energy levels
(Spacingr
l
~---1f----,rJn;:
3
---~~2~!i;
~--+--~
II
z;
2 ---'--1
(a)
,..-"--,
N-fold
degenerate
levels
(b)
Bands.
each
with
N values
of
k
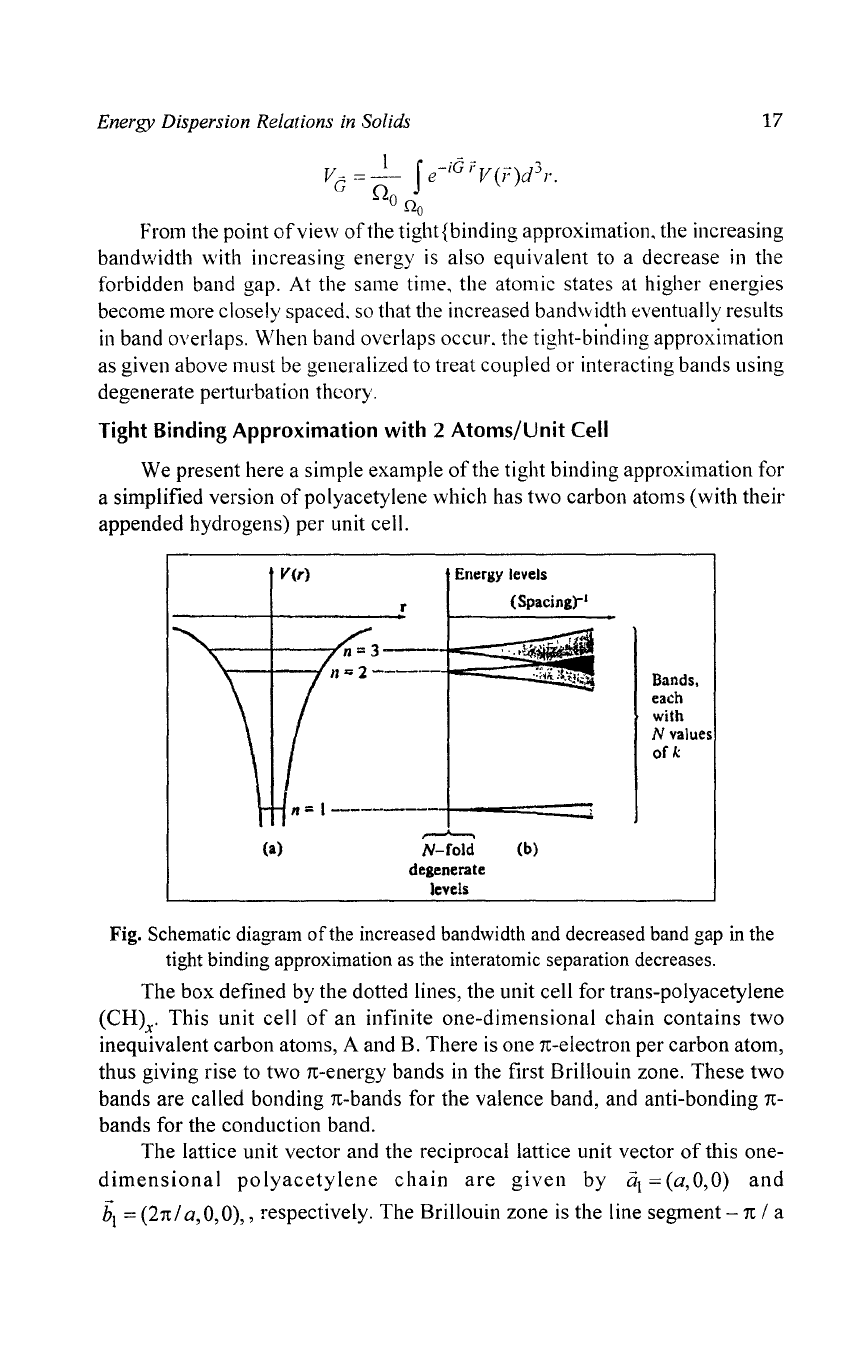
Fig. Schematic diagram
of
the increased bandwidth and decreased band gap
in
the
tight binding approximation as the interatomic separation decreases.
The
box
defined by the dotted lines, the unit cell for trans-polyacetylene
(CH)x'
This
unit
cell
of
an
infinite
one-dimensional
chain
contains
two
inequivalent carbon
at0111s,
A and B. There
is
one
TC-electron
per carbon atom,
thus giving rise to
two
TC-energy
bands in the first Brillouin zone. These
two
bands are called bonding
TC-bands
for the valence band, and anti-bonding
TC-
bands for the conduction band.
The lattice unit vector and the reciprocal lattice unit vector
of
this one-
dimensional
polyacetylene
chain
are
given
by
al
=(a,O,O)
and
b
l
=
(21t1
a,
0,
0)"
respectively.
The
Brillouin zone
is
the line segment -
TC
I a

18
Energy Dispersion Relations in Solids
< k < 1t /
a.
The
Bloch
orbitals consisting
of
A and B atoms are given
by
1 "
'kR
y}
(r) =
r;:;
L.
el
a~}
(r - Ra),(·,x =
A,B)
'\IN
R
a
where
the summation is taken over the atom site coordinate
Ra.
for the A
or
B carbon ahmlS
in
the solid.
To solve for
the
energy
eigenvalues and \\avefullctions we need to solve
the general equation:
H()=
ES~'
where
H
is
the
17
x
11
tight binding matrix Hamiltonian for
the
17
coupled
bands
(ll = 2
in
the
case
of
polyacetylene) and S is
the
corresponding
11
x
11
overlap integral matrix.
To
obtain a solution to this matrix equation, we require
that
the determinant
IH
-
ESI
vanish.
This approach is
easily
generalized to periodic structures with more than
2 atoms
per
unit cell.
The
(2 x 2)
matrix
Hamiltonian,
Ha.~'
(a,
~
= A, B) is
obtained
by
substituting equation
H}/(k)
=
(tfl)
I
HI
tfl}'
),S}}'(k) =
(tfl)
I
tfl]'
)(J,j'
= 1,2),
r~"""''''!H
iii
I
i c i c
,
/.(A)
'\.,!B)
/.
" /
c ,
c,
c
I
ill
I
H i H 1 H
~
.............
.:
Fig. The unit cell oftrans- polyacetylene bounded by a box defined by the dotted
lines, and showing two inequiva-Ient carbon atoms, A and
S,
in the unit cell.
- -
where
the
integrals
over
the
Bloch
orbitals,
Hj]'(k)
and
Sjj'(k)"
are
called
transfer
integral matrices
and
overlap
integral matrices, respectively.
When
a =
~
= A,
we
obtain
the diagonal
matrix
element
H (r) =
J.-
I
eik(R-R')
($
A(r - R') I H I
~
A(r -
R»)
AA
N R,R'
J.-
IE
2P
+J.-
I
e±lka(~A(r-R')IHI$A(r-R»)
N R,R' N R'=R±a
+(terms
equal
to
or
more
distant
than
R'=
R ± 2a).
= E
2p
+ (terms equal
to
or
more
distant
than
R'
= R ±a).
The
main
contribution
to
the
matrix
element
HAA
comes
from
R'=
R,
and
this gives
the
orbital
energy
of
the
2p level, E
2p
'
We
note
that
E
2p
is not simply
the
atomic
energy
value for the free atom,
because
the
Hamiltonian
H also

Energy Dispersion Relations in Solids
19
includes a crystal potential contribution. The next order contribution to HAA
in
equation comes from terms
in
R'
= R ± (/, which are here neglected for
simplicity. Similarly,
HBB
also gives E
2p
to the same order
of
approximation.
Next
let us consider the off-diagonal matrix element
H.w
(r)
which
explicitly couples the
A unit to the B unit. The largest contribution to
~~B
(r)
arises when atoms A and B are nearest neighbors. Thus
in
the sLlmmation over
R', we only consider the terms with R' = R ± a
12
as a first approximation and
neglect more distant terms to obtain
1
",r
-rka/l(
)
Hw(r)=
NL....(e -
~A(r-R)IHI~B(r-R-a!2)
R
+
e-
rka
12
(~
A
(r
- R) I H I
~
B
(r
- R - a ! 2)
)}
=
2t
cos(ka!2)
where
t
is
the transfer integral appearing
in
equation and
is
denoted by
t =
(~A
(r
- R) I H I
~
B
(r
- R ±
a!
2»)
.
Here we have assumed that all the 1t bonding orbitals are
of
equal length
(1.5°A bonds). In the real
(CH)x compound, bond alternation occurs, in which
the bonding between adjacent carbon atoms alternates between single bonds
(1. 7
A) and double bonds (1.3° A). With this bond alternation, the two matrix
elements between atomic wavefunctions in equation are not equal. Although
the distortion
of
the lattice lowers the total energy, the electronic energy always
decreases more than the lattice energy in a one-dimensional material. This
distortion deforms the lattice by a process called the Peierls instability. This
instability arises for example when a distortion is introduced into a system
containing a previously degenerate system with 2 equivalent atoms per unit
cell. The distortion making the atoms inequivalent increases the unit
cell by a
factor
of
2 and decreases the reciprocal lattice by a factor
of
2.
If
the energy
band was
formally
half
filled, a band gap
is
introduced by the Peierls instability
at the Fermi level, which lowers the total energy
of
the system.
It
is
stressed
that t has a negative value. The matrix element
HBir)
is obtained from H
AB
-
(r)
through the Hermitian conjugation relation HBA =
If
AB' but since
~~B
is
real, we obtai
H
BA
= H
AB
·
The overlap matrix
SIj
can be calculated by a similar method as was used
for
Hi)' except that the intra-atomic integral
Si)
yields a unit matrix
in
the limit
of
large in-teratomic distances,
if
we assume that the atomic wavefunction
is
normalized so that SAA = S
BB
= I. It
is
assumed that for polyacetylene the SAA
and
SBB
matrix elements are still approxi-mately unity. For the off-diagonal
matrix element for polyacetylene we have
SAB
= SBA = 2s cos(ka!2), where s
is
an overlap integral between the nearest A and B atoms,
20
Energy Dispersion Relations
in
Solids
s =
($
A(r
- R) I
$B(r
-R
±
aI2)).
The secular equation for the
2p:;
orbital
of
CHI is obtained by setting he
determinant
of
E)
-E
-p
2(t
- sE)cos(ka 12)
2(1
- sE)cos(ka I
2)
E
2p
- E
= (E
2p
-
E)2
-
4(t
-
sE?
cos
2
(kaI2)
=0
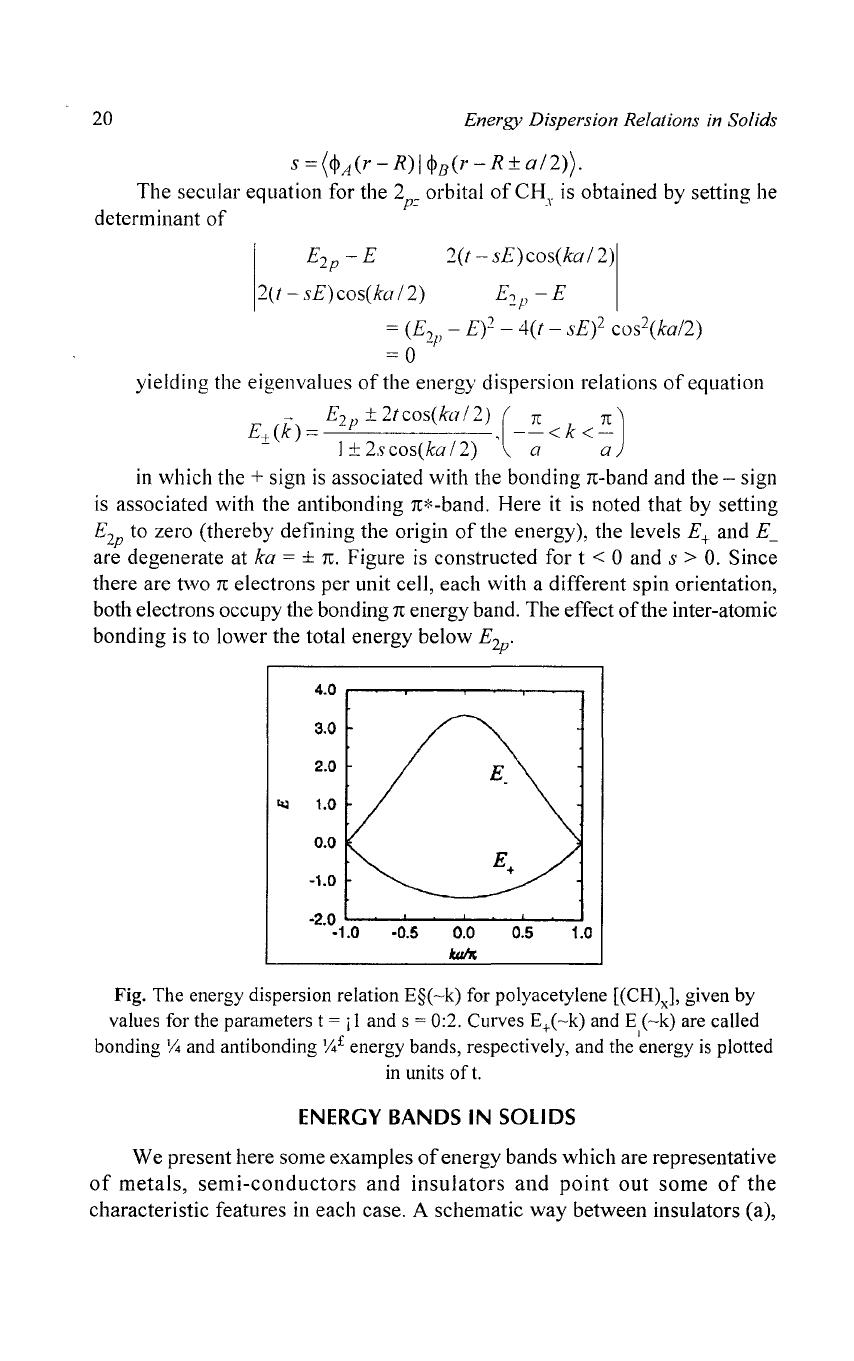
yielding the eigenvalues
of
the energy dispersion relations
of
equation
~
E
2p
± 2tcos(kaI2)
(IT
IT)
E+
(k)
= ,
--
< k
<-
~
1±2scos(kaI2)
a a
in
which the + sign
is
associated with the bonding
IT-band
and the - sign
is
associated with the anti bonding IT*-band. Here it
is
noted that by setting
E
2p
to zero (thereby defining the origin
of
the energy), the levels
E+
and
E~
are degenerate at ka = ±
TC.
Figure
is
constructed for t < 0 and s >
O.
Since
there are two
TC
electrons per unit cell, each with a different spin orientation,
both electrons occupy the bonding
TC
energy band. The effect
of
the inter-atomic
bonding is to lower the total energy below
E
2p
'
4.0
3.0
2.0
~
1.0
0.0
E+
-1.0
-2.0
-1.0 -0.5
0.0
0.5
1.0
kuIIt
Fig. The energy dispersion relation
E§(-k)
for polyacetylene
[(CH)J,
given by
values for the parameters
t = j I and s = 0:2. Curves E+(-k) and E
(-k)
are called
I
bonding
Y4
and anti bonding
Y4£
energy bands, respectively, and the energy
is
plotted
in units
of
t.
ENERGY
BANDS
IN
SOLIDS
We
present here some examples
of
energy bands which are representative
of
metals,
semi-conductors
and
insulators
and
point
out
some
of
the
characteristic features
in
each case. A schematic way between insulators (a),

Energy Dispersion Relations
in
Solids 21
metals (b),
semimetals
(c), a
thermally
excited
semiconductor
(d) for
which
at
T=
0 all states in
the
valence band are occupied and all states
in
the conduction
band are
unoccupied
,
assuming
no impurities or crystal defects. Finally,
we
see a
p-doped
semiconductor
which
is
deficient
in
electrons,
not
having
sufficient
electrons
to fill the valence band
complet
ely as in
(d)
.
The
electron dispersion relations for an
in
sulator (a), while (c)
shows
dispersion relations for a metal. A semimetal
if
the
number
of
e:ectrons
equals
the
number
of
holes,
but
a metal otherwise.
METALS
Alkali Metals-e.g.,
Sodium
For
the
alkali
metals
the
valence
electrons
are nearly free and the weak
binding approxil1la-tion describes these electrons quite well.
The
Fermi surface
is
nearly spherical and the band gaps
are
small.
The
crystal structure for the
alkali
metals
IS
body
centered
cubic
(Bee)
and
the
E(k)
diagram is
drawn
starting with the bottom
of
the
half-filled
conduction
band.
For
example,
the
E(k)
for
sodium
begins
at
-
-0.6
Rydberg
and
represents the
3s
conduction
band
.
The
filled
valence
bands
lie much lower in
energy.
For
the
case
of
sodium, the 3s conduction band
is
very
nearly free electron
like and
the
E(k)
relations are
closely
isotropic.
Thus
the
E(k)
relations
along
the
~(1
00), )2(
11
0) and A(
Ill)
directions
are
essentially
coincident
and
can be so plotted.
For
these
metals, the
Fermi
level is
determined
so
that
the
3s
band
is
exactly
half-occupied,
since
the
Brillouin
zone
is large
enough
to
accommodate
2
electrons
per
unit cell.
Thus
the radius
of
the
Fermi
surface
kF
satisfies the relation
where
V
B
2.
and a are, respectively, the
volume
of
the
Brillouin
zone
and
the
lattice constant.
4 k
3
1 I
(211:)3
kFa
..,
-11:
F=-V
BZ
=-(2)
-
.or-~O.6.J
,
3 2
··
2 a
211:
For
the
alkali metals, the effective mass
III
*
is
nearly equal to
the
free
electron mass
111
and
the
Fermi surface is nearly spherical and
never
comes
close to the Brillouin zone boundary.
The
zone
boundary
for the )2, A and
~
directions
are
indicated
in
the
E(k)
by vertical lines.
For
the alkali metals
the band gaps are
very
small
compared
to the band
widths
and the
E(k)
relations
are
parabolic
(E
= h
2
k
2
12m*)
almost
up to
the
Brillouin
zone
boundaries. By
comparing
E(k)
for
Na
with
the
Bee
empty
lattice bands for