Мамаев Н.Н. Гематология: руководство для врачей
Подождите немного. Документ загружается.


Виллебранда (достаточно тяжелых формах),
при
афибриногенемии, дефицита!
факторов
VII и X,
ДВС-синдроме
и
диспротеинемиях. Тип кровоточивости опре
деляется
на
основании оценки
ее
клинических проявлений
—
главным образом
по
характеру кровотечения при повреждении сосудов разного калибра
и по прв
обладающей локализации геморрагии.
При
капиллярном типе нарушения целостности стенок капилляров
и
ар тс
риол поверхностные порезы сопровождаются продолжительным кровотечени-
ем,
которое начинается сразу после травмы,
так
как гемостаз
в
этих
сосудах
осу
ществляется
за
счет сосудисто-тромбоцитарных реакций. Поскольку при коагу
ляционном
типе сосудистый
и
тромбоцитарный компоненты гемостаз^!
существенно
не
изменены, сразу после травмы кровотечений,
как
правило,
не
бывает. После повреждения сосудов среднего калибра
при
таких операциях,
как
удаление зубов, тонзиллэктомия, кровотечение
у
больных
с
коагуляцион-
ным
дефектом вначале останавливается
за
счет образования первичной тромбо-
цитарной
пробки
и
сосудистых реакций. Однако через
2
часа
(и
более) после
восстановления кровяного давления
в
сосудах
травмированной области оно воз-
обновляется,
так
как при коагулопатиях нарушено образование фибрина
и
соот-
ветственно тромбоцитарно-фибринового тромба, способного противостоять
давлению крови
и
обеспечить окончательный гемостаз
в
сосудах
среднего
диа-
метра.
У
больных
с
дефектами
в
тромбоцитарном звене гемостаза кровотечение
после повреждения сосудов среднего диаметра также усилено,
но, в
отличие
от
пациентов
с
коагуляционной патологией,
у них
после операции
не
наблюдается
временной
остановки кровотечения.
Это
объясняется тем,
что при
тромбоцито-
пении
и
тромбоцитопатиях изменен
как
первичный гемостаз из-за дефекта
тромбоцитарного звена гемостаза,
так и
вторичный вследствие затруднения
об-
разования
фибринового сгустка из-за недостаточного участия кровяных пласти-
нок
в
коагуляционном процессе.
Каждый
тип
кровоточивости характеризуется определенной локализацией
геморрагии (табл.
3.2).
Так, петехии, поверхностные множественные синяки
и,
прежде всего, кровотечения
из
слизистых оболочек (носовые, маточные,
желу-
дочно-кишечные) характерны
для
со
суди
сто-тромбоцитарных заболеваний,
в то
время
как
глубокие гематомы
— для
коагуляционных нарушений,
а
гемартро-
зы
—
для
тяжелых форм последних.
Определение типа кровоточивости содействует
не
только установлению
ос-
новного
гемостатического нарушения,
но и
планированию дальнейшего лабора-
торного обследования больного. Так, при капиллярном типе кровоточивости не-
обходимо больше внимания
уделить
оценке состояния сосудисто -тромбоцитар-
ного звена гемостаза,
при
коагуляционном
—
плазменно-коагуляционного,
но
никогда
нельзя ограничиваться
только одним направлением исследования.
Б
любом
случае
должны быть выполнены скрининговые (общие) тесты, харак-
теризующие
все
компоненты системы гемостаза (табл.
3.3). Это
обусловлено
возможностью существования вышеуказанных смешанных форм нарушений
в
нескольких звеньях этой системы. Особенно часто встречаются комбиниро-
ванные
дефекты
при
приобретенных формах кровоточивости,
при
которых
не-
редко имеются изменения
в
нескольких звеньях системы гемостаза.
Рационально
оценивать геморрагические проявления
по
степени тяжести,
что имеет существенное значение
для
оценки трудоспособности больного.
При
этом
следует
учитывать
как
тяжесть геморрагических эпизодов,
так и их
частоту,

•
МИШИН)!
И
I1IUII)[)IIM1|)MI1H
ДИ.1ИШЫИМ WMU\)\JM И-ИНКИД ДН
1
Таблица 3.2
Определение
характера
кровоточивости по клиническим проявлениям
" икис проиндвния
тматомы
in кровотечения
•*••
м;л"чно-кишвчные
чония
,'|>ИЯ
M«t ii.io кровотечения
мочения после
• ции зубов
чтрационные
"1"ч..рм>чения
шрные проявления
tint
уморенных формах
Характер кровоточивости
Коагуляционный
Большие
Часто встречаются у тяжелых
больных как основной признак
Наблюдаются редко
Как
преобладающий синдром
встречаются редко, пока нет
изъязвления
Характерна
Не характерны (большинство
больных — мужчины)
Начинаются через несколько
часов после операции и не
останавливаются после нало-
жения давящей повязки
Поздние кровотечения с обра-
зованием раневой гематомы
Большие гематомы после трав-
мы и опасные кровотечения по-
сле ранений
Капиллярный
Небольшие, поверхностные
Не характерны
Часто основной вид кровотече-
ний
Преобладающий синдром
Не характерна
Характерны
Начинаются сразу после опера-
ции
и обычно останавливаются
после наложения давящей по-
вязки
Кровотечения в основном во
время операции
Носовые и маточные кровоте-
чения
Таблица 3.3
Обязательный объем лабораторных исследований при первичном обследовании
больного
с
геморрагическим
диатезом
Тесты для характеристики
плазме нно-коагуляционного звена гемостаза
1 Нрпмя свертывания венозной крови
1 Лмивированмое парциальное тромбопластиновое
IplMfl
i Мротромбиновый тест
А 1ромбиновое время
ицентрация фибриногена
|
1
|С1 растворимости фибринового сгустка
•пине
(активность фактора
XIII)
Тесты для характеристики
сосудисто-тромбоцитарного
эвена гемостаза
1.
Резистентность сосудистой стенки
{тест
жгута)
2. Количество тромбоцитов
3.
Длительность кровотечения
i
ршкже
локализацию
геморрагии
и
качество
жизни пациента (см.
карту
опроса).
<'единение
этих
признаков
позволяет
дать
оценку
степеней
тяжести,
которая
||||>н()дится
раздельно
для капиллярного и
коагуляционного
типов кровоточи-
ПМ
111.
I
степень
—
петехии,
небольшие
экхимозы,
умеренные
посттравматические
•рютечения
больные
ведут
нормальный
образ
жизни.
II
степень
— кровотечения из
слизистых
оболочек
средней
тяжести,
боль-
iiMi'
жхимозы,
умеренные
гематомы,
быстро
преходящие
гемартрозы;
геморра-
ii'in
кие эпизоды
возникают
почти
ежегодно.

I 11,1 П.! 1 I СМ1>|1|1.!1ИЧШ КИГ
III
степень —
тяжелые кровотечения
из
слизистых оболочек, приводящие
к
госпитализации, тяжелые гемартрозы, кровоизлияния
во
внутренние органы;
ре
моррагические эпизоды наблюдаются
2—5 раз
в
год;
трудоспособность сущее
т
венно
ограничена.
IV степень —
угрожающие жизни кровотечения различной локализации;
ге-
моррагические эпизоды возникают почти ежемесячно; трудоспособность боль-
ного полностью нарушена.
На
основании совокупного анализа
могут
быть поставлены следующие
во-
просы.
Первый: является
ли
кровоточивость
у
больного следствием нарушений
в
системе гемостаза
или она
связана
с
местными сосудисто-тканевыми измене-
ниями?
Второй: если имеются изменения
в
системе гемостаза,
то
являются
ли
они
врожденными
или
приобретенными? Третий: каков
тип
кровоточивости?
Четвертый: какова степень тяжести геморрагических проявлений? Оценивать
анамнестические сведения нужно
в
совокупности,
так как
отдельно взятые
при-
знаки
имеют относительное значение. Наиболее сложно ответить
на
первый
во-
прос,
для
этого требуется анализ многих данных
о
течении заболевания,
в
том
числе ответы
на
второй
и
третий вопросы,
и
нередко
—
дополнительное обсле-
дование больного
у
различных специалистов,
а
также исследование системы
ге-
мостаза
у
кровных родственников пациента.
Лабораторная диагностика
нарушений
сосудисто-тромбоцитарного
звена
гемостаза
В
процессе диагностики тромбоцитопатий
с
целью установления недостаточ-
ности
функциональной активности тромбоцитов
и
определения механизма
ее
развития исследования проводятся
в
три
этапа, которые связаны
и с
возможно-
стями отдельных медицинских учреждений (табл.
3.4).
I этап —
установление факта нарушений тромбоцитарного звена гемостаза.
Данный
этап
в
основном должен выполняться всеми медицинскими
учрежде-
ниями,
в
том
числе
и
самого нижнего уровня
—
поликлиниками, участковыми
службами.
II
этап —
определение фазы тромбоцитарных превращений,
в
которой имеет-
ся
доминирующее нарушение.
Это
осуществляется
на
основании выполнения
комплекса
функциональных реакций, анализ совокупности которых достаточен
для установления диагноза основных форм тромбоцитарных заболеваний
и
синд-
ромов.
Данные исследования выполняются
в
гемостазиологических лабораториях
крупных стационаров
и
поликлиник,
где
предусмотрена работа гематолога.
III
этап —
определение структурно-молекулярных
и
генетических дефектов,
лежащих
в
основе изменения функции сосудисто-тромбоцитарного звена гемо-
стаза. Решение этой задачи доступно специализированным научным лаборато-
риям,
но
характер
и
направление этих исследований определяются результата-
ми,
полученными
на
предыдущих этапах.
Как
видно
из
приведенного алгоритма, первый этап осуществляется
на
осно-
вании
анализа данных анамнеза, первичного осмотра больного,
а
также
по ре-
зультатам ориентировочных (скрининговых) тестов первичного гемостаза
—
длительности кровотечения, количества тромбоцитов
и,
возможно, адгезив-
но-агрегационных тестов.
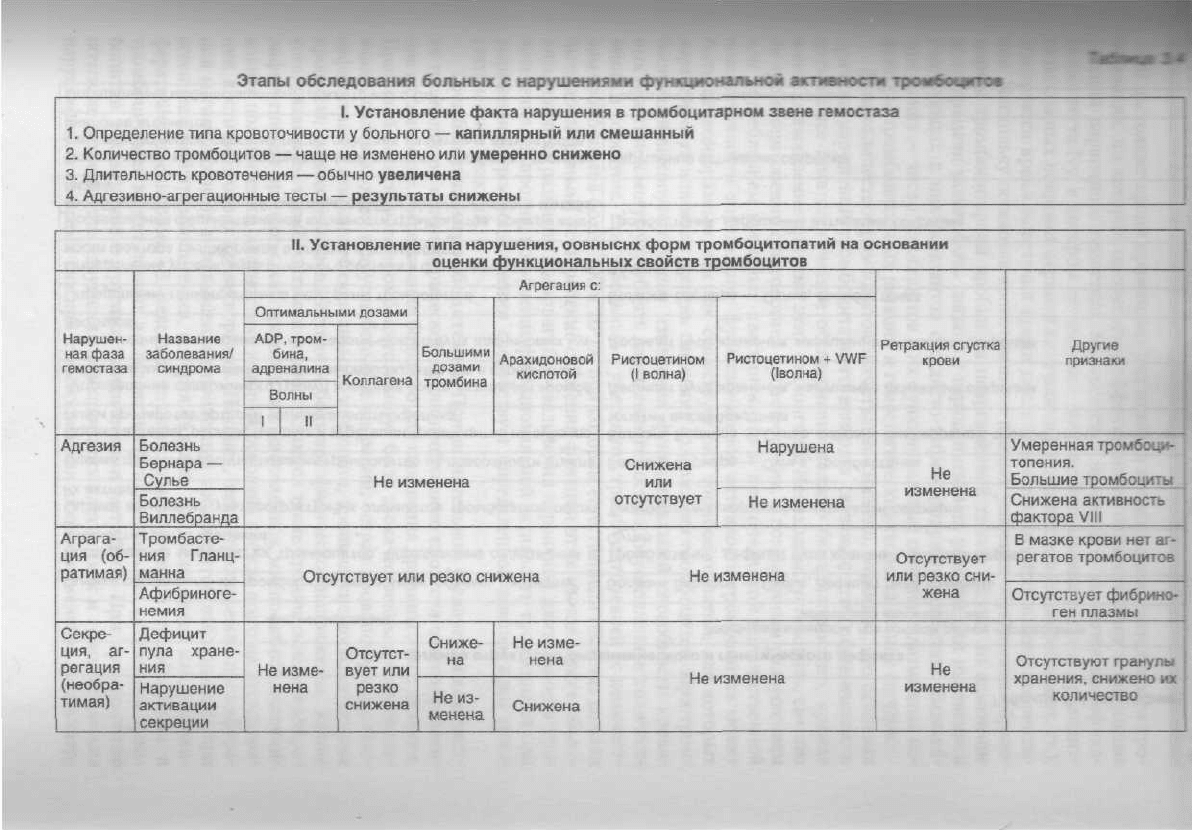
Этапы
обследования
больных
с нарушениями функциональной активности тромбоцитоа
I. Установление
факта
нарушения в тромбоцитарном звене гемостаза
1.
Определение типа кровоточивости у больного — капиллярный или смешанный
2. Количество тромбоцитов —
чаще
не изменено или умеренно снижено
3. Длительность кровотечения — обычно
увеличена
4.
Адгезивно-агрегационные тесты — результаты снижены
II.
Установление типа нарушения, оовныснх форм тромбоцитопатии на основании
оценки
функциональных
свойств тромбоцитов
Нарушен-
ная
фаза
гемостаза
Адгезия
Аграга-
ция (об-
ратимая)
Секре-
ция,
аг-
регация
(необра-
тимая)
Название
заболевания/
синдрома
Болезнь
Бернара —
Сулье
Болезнь
Виллебранда
Тромбасте-
ния Гланц-
манна
Афибриноге-
немия
Дефицит
пула
хране-
ния
Нарушение
активации
секреции
Агрегация с:
Оптимальными дозами
ADP,
тром-
бина,
адреналина
Волны
I И
Коллагена
Большими
дозами
тромбина
Арахидоновой
кислотой
Не изменена
Отсутствует или резко снижена
Не
изме-
нена
Отсутст-
вует или
резко
снижена
Сниже-
на
Не из-
менена
Не
изме-
нена
Снижена
Ристоцетином
(I
волна)
Снижена
или
отсутствует
Ристоцетином + VWF
(1волна)
Нарушена
Не изменена
Не изменена
Не изменена
Ретракция сгустка
крови
Не
изменена
Отсутствует
или резко сни-
жена
Не
изменена
Другие
признаки
Умеренная тромбоци-
ТОПвНЙЯ.
Большие тромбоциты
Снижена активность
фактора VII!
В мазке крови нет аг-
регатов тромбоцитов
Отсутствует фибрино-
ген
плазмы
Отсутствуют гранулы
хранения, снижено их
количество
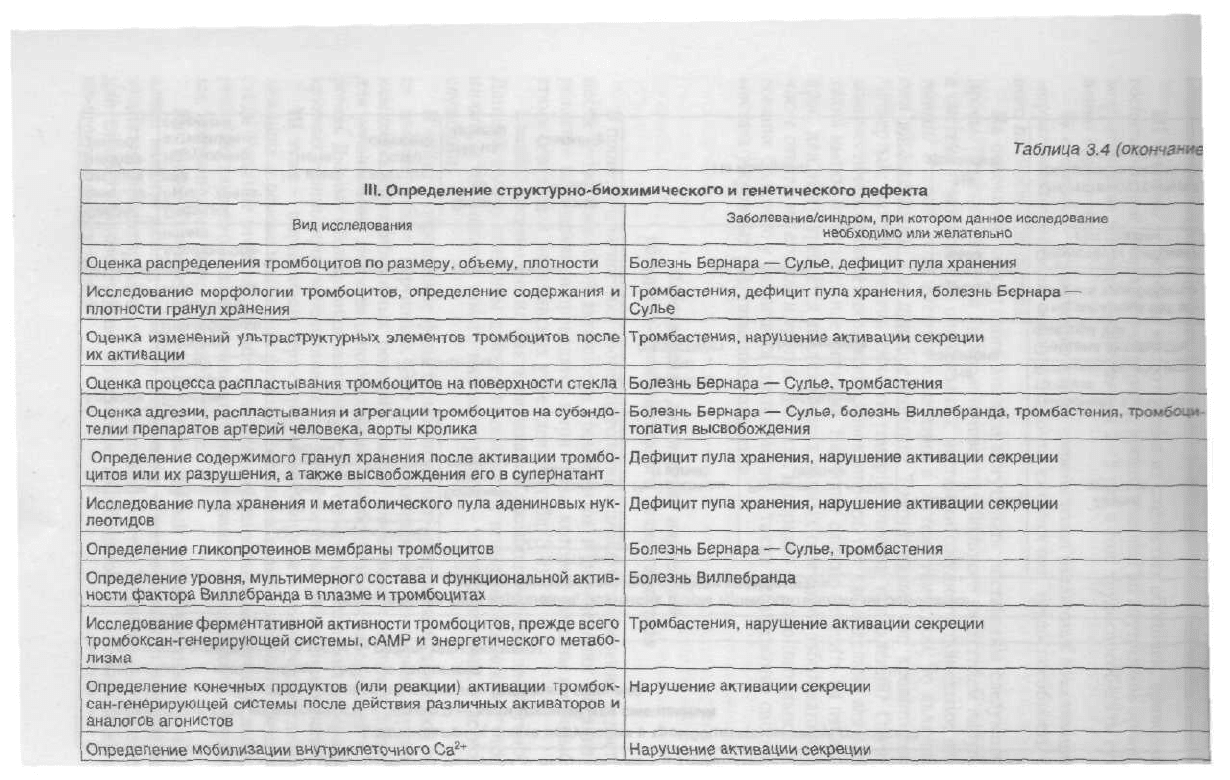
Таблица
3.4
(окончание
I.
Определение структурно-биохимического и генетического дефекта
Вид
исследования
Заболевание/синдром,
при котором данное исследование
необходимо или
желательно
Оценка распределения тромбоцитов по размеру, объему, плотности
Болезнь Бернара —
Сулье,
дефицит
пула
хранения
Исследование морфологии тромбоцитов, определение содержания и
плотности гранул хранения ^^^
Тромбастения, дефицит
пула
хранения, болезнь Бернара —
Сулье
Оценка изменений ультраструктурных элементов тромбоцитов после
их активации
Тромбастения, нарушение активации секреции
Оценка процесса распластывания тромбоцитов на поверхности стекла Болезнь Бернара —
Сулье,
тромбастения
Оценка
адгезии,
распластывания и агрегации тромбоцитов на субэндо-
телии препаратов артерий человека, аорты кролика
Болезнь Бернара —
Сулье,
болезнь Виплебранда, тромбастения, тромбоци-
топатия высвобождения
Определение содержимого гранул хранения после активации тромбо-
цитов или их разрушения, а также высвобождения его в супернатант
Дефицит
пула
хранения, нарушение активации секреции
Исследование
пула
хранения и метаболического
пула
адениновых нук-
леотидов
Дефицит пупа хранения, нарушение активации секреции
Определение гликопротеинов мембраны тромбоцитов Болезнь Бернара —
Сулье,
тромбастения
Определение уровня, мультимерного состава и функциональной актив-
ности
фактора Виллебранда в плазме и тромбоцитах
Болезнь Виллебранда
Исследование ферментативной активности тромбоцитов, прежде всего
тромбоксан-генерирующей
системы, сАМР и энергетического метабо-
лизма
Тромбастения, нарушение активации секреции
Определение конечных продуктов (или реакции) активации тромбок-
сан-генерирующей системы после действия различных активаторов и
аналогов агонистов
Нарушение активации секреции
Определение мобилизации внутриклеточного Са
2
Нарушение активации секреции

тшцсс иреми применяют ряд методов исследования длительности кро-
II».
ш) которой оценивается эффективность осуществления в организме
первичного гемостаза. Большинство из методов являются модифика-
• i m Ivy, в котором стандартное повреждение мелких
сосудов
проводит-
1ЖИЫХ покровах верхней конечности в условиях веностаза (40 мм рт. ст.).
пи
время истечения крови после нанесения дозированного поврежде-
I
lldi
кольку остановка кровотечения в данных условиях обусловлена образо-
фпмГюцитарной пробки на поврежденной стенке
сосуда,
то изменение
" времени может быть связано с нарушением
всех
реакций или изменением
1мцих в них реактантов сосудисто-тромбоцитарного гемостаза. Важней-
и
i них число тромбоцитов и их функциональные свойства, плазменные ко-
i
тромбоцитарных реакций и гемостатические свойства стенок сосудов.
11 in количество тромбоцитов значительно снижено, очевидно, что основная
||'м мим нарушений тромбоцитарного аппарата связана с этим изменением, и
it
iiriiiiwc
гематологическое обследование
должно
быть направлено, прежде
м.1 появление причины этих отклонений. Необходимо установить, связана
чмГюцитопения с патологией кроветворения, с повышенным разрушением
потреблением кровяных пластинок. Но и при очевидной количественной
Ни
м сочности определение функциональной активности тромбоцитов может
целесообразным, так как
существует
ряд тромбоцитопатий, для которых
i
|" горна та или иная степень снижения содержания тромбоцитов. При тром-
пении
сопутствующие изменения функциональной способности кровя-
1
1.КТИНОК
могут
усугублять
нарушение гемостаза, обусловленное количест-
i.iM дефектом. Так, у больных с умеренной тромбоцитопенией (с количест-
Фомбоцитов более 100 х
10
9
/л,
при котором естественный гемостаз должен
i
достаточным) установление снижения функции этих клеток может объяс-
ним,
причину развития геморрагии и увеличения первичной длительности кро-
кмия.
Напротив, повышение функциональной активности тромбоцитов при
• цмженной тромбоцитопении способствует определенной компенсации гемо-
нческих реакций.
I
|м>мбоцитоз и тромбоцитемия. При значительном увеличении количест-
!'омбоцитов в периферической крови необходимо провести целенаправлен-
/ншическое обследование больного для выяснения причины этого явления.
Причиной
реактивного тромбоцитоза
могут
быть злокачественные новообразо-
|. гпленэктомия и
другие
большие операции, острая кровопотеря, железо-
щитная
и гемолитическая анемии,
инфекции,
хронические воспалительные
Процессы,
стрессорные воздействия и большое физическое напряжение. Кроме
п>П),
развитием тромбоцитемии сопровождаются такие хронические миелопро-
||цфсративные процессы, как хронический миелолейкоз, миелофиброз, эссенци-
i
ш полицитемия, диагностика которых основана прежде всего на исследова-
нии
костномозгового кроветворения. Наконец, неконтролируемая пролифера-
ции
мегакариоцитов и повышенная продукция тромбоцитов наблюдаются при
и
i социальной тромбоцитемии, которая является одним из видов миелопроли-
'|м рлтивного синдрома. В неясных
случаях
разграничению тромбоцитоза и
|рс1мГюцитемии может помочь исследование функциональной активности тром-
ппцитов в комплексе с другими клиническими методами. При тромбоцитозе
почти не встречаются нарушения функции этих клеток (как и гемостатические
и
(рушения), в то время как при тромбоцитемии данные изменения закономер-

ны
(у больных нередко наблюдаются геморрагические проявления или тромбя
зы,
a in
vitro
после стандартизации содержания пластинок в богатой тромбо|Я
тами плазме часто выявляют снижение функциональных свойств этих клетокЩ
Наличие
кровоточивости капиллярного типа, увеличенная первичная дл|
тельность кровотечения при нормальном количестве тромбоцитов в периферЛ
ческой крови указывают на возможность функциональных нарушений в
сосудв
сто-тромбоцитарных реакциях.
После
установления нарушений в тромбоцитарном звене гемостаза
следув
приступить ко второму этапу исследования с целью определения характер
тромбоцитопатии. Анализ результатов относительно небольшого комплекса мщ
тодов
дает
возможность дифференцировать основные формы нарушений
сосщ
дисто-тромбоцитарного гемостаза, обусловленные как собственно тромбоцитнрч
ным
дефектом, так и недостаточностью плазменных кофакторов первичного Д
мостаза (см. табл. 3.4).
В данном комплексе методов главной является оценка агрегационного npoJ
цесса тромбоцитов с помощью приборов, основанных на фотометрическом]
принципе.
Агрегация тромбоцитов в этих агрегометрах оценивается путем ввм
дения
в кювету прибора стабилизированной плазмы больного со стандартной
ванным
до определенного уровня количеством тромбоцитов, где она перемеши-1
вается с одним из естественных агонистов определенной концентрации. Быстро!
образующиеся в этих условиях тромбоцитарные агрегаты во времени увеличн»!
ваются в количестве и размере, и это коррелирует с повышением степени прохов
ждения через плазму
луча
света, поскольку в
результате
включения тромбоци-1
тов в агрегаты она становится менее плотной. Поэтому степень повышения про-1
хождения светового
луча
служит мерой оценки агрегационного процесса.)
Применение
различных агрегирующих агентов позволяет с каждым из них оце-
нивать нарушения, в основе которых
могут
лежать изменения в разных фазах
активации
тромбоцитов (связь естественного агониста с рецептором тромбоци-
тов, передача сигналов активации, стимуляция финальных реакций клетки).
Первая
волна агрегации соответствует фазе обратимой агрегации, а вторая,
развивающаяся после латентного периода, вызвана эндогенными агрегирующи-
ми
агентами, секретируемыми в это время из гранул хранения тромбоцитов. По-
этому величина второй волны может характеризовать как реакции активации i
секреторного процесса, так и количество содержащихся в гранулах агрегирую-
щих агентов. В отличие от естественных агонистов, агрегация с ристомицином
воссоздает механизм адгезивного процесса, поскольку этот антибиотик изменяет
конформацию
фактора Виллебранда и тромбоцитарного рецептора GPIb-IX-V,
;
подобно тому как это происходит при контакте данных молекул с адгезивными
:
субэндотелиальными структурами в условиях высокой скорости тока крови при
повреждении стенки сосудов микроциркуляции.
В настоящее время установлено, что геморрагические проявления при врож-
денной
или приобретенной недостаточности тромбоцитарного звена гемостаза
могут
быть обусловлены тем, что у больного имеются нарушения на той или
иной
ступени последовательно развивающихся гемостатических реакций кровя-
ных пластинок: адгезии, агрегации, секреции, реализации коагуляционной ак-
тивности кровяных пластинок и ретракции гемостатической пробки.
Нарушение в фазе адгезии устанавливают на основании неизмененных аг-
регационных реакций со всеми растворимыми естественными агонистами и от-

ним
резкого снижении первой
волны
агрегации
с
ристоцетином, кото-
1Ктгризует адгезивный процесс. Если последний дефект корригируется
мнем
it
кювету агрегометра фактора Виллебранда, содержащегося
в бес-
ммнпиоп
плазме здоровых людей,
то
можно
думать,
что
недостаточность
|.|||||ц|()
гемостаза, которая проявилась
в
возникновении геморрагии
и
ими
типичной длительности кровотечения, обусловлена снижением
со-
•
i (ини
изменением функциональных свойств) этого плазменного
ко-
щпчии.
Такое нарушение наблюдается, прежде всего,
при
болезни
Вил-
i
\л ни
подобной коррекции
нет, то это
свидетельствует,
что
дефект
in
гимн тромбоцитов
и
фактора Виллебранда
в
присутствии ристоцети-
11
!
обственно тромбоцитарным дефектом
— с
отсутствием
на
мембране
гик
i
ликопротеинового рецептора
для
фактора Виллебранда, находяще-
илексе GPIb-IX-V,
т. е. с
болезнью Бернара
—
Сулье.
II 1><
(положение
о
болезни Виллебраида подтверждается аутосомно-доми-
..фпктером наследования
и
сочетанием выявленного дефекта
в
фазе
•
I
снижением коагуляционной активности фактора VIII, поскольку
фак-
1г(>|мнда является носителем молекулы фактора VIII
и
защищает
по-
i
мт
разрушения
и
элиминации
в
циркулирующей крови.
У
пациентов
иной
или
тяжелой формой болезни Виллебранда кровоточивость
мо-
11.
не
капиллярного,
а
смешанного типа.
'I
i
болезни Бернара
—
Сулье
подтверждается аутосомно-рецессив-
мм
i
щи
обом наследования
и
обнаружением
в
мазке крови гигантских тромбо-
(диаметром более
4
мкм). Обычно
у
этих больных имеется
и
умеренная
пцитопения.
IT
отметить,
что
морфологические особенности тромбоцитов
в
мазке
«.ни
могут
представлять ценную информацию
и при
диагностике
других
тром-
1О1ШТИЙ.
Большие тромбоциты наблюдаются также
при
иммунной тромбо-
1'пии,
синдроме серых тромбоцитов, аномалии
Мея —
Хегглина; напротив,
и'1> уменьшен
при
синдроме Вискотта
—
Олдрича; сниженное гранулиро-
кроияных
пластинок выражено
при
синдроме серых тромбоцитов, ос,
6-де-
iMtnne
пула
хранения; почти полное
отсутствие
в
мазке периферической крови
пиарных агрегатов
и
отростков
у
этих клеток
— при
тромбастении
•мм.шпа.
Следующие
два
основных заболевания
с
нарушениями
в
фазе обратимой
•
-тми, как и
предыдущая группа заболеваний, также обусловлены
или соб-
тромбоцитарным мембранным дефектом
или
отсутствием/дефектом
lennoio кофактора агрегации,
т. е.
фибриногена.
О
недостаточности
агрега-
N
'О
процесса
свидетельствует
резкое снижение
или
даже
отсутствие
пер-
•ггорой волны агрегации
в
богатой тромбоцитами плазме
с
оптимальными
•
Ими АДФ и
других
естественных растворимых агонистов,
а
также единствен-
i
олны
с
умеренными дозами коллагена. Такой
тип
изменений характерен
•
рожденного заболевания
с
аутосомно-рецессивным способом наследова-
тромбастении
Гланцманна, при
которой
в
мембране тромбоцитов
от-
шутт,
резко снижены
в
количественном отношении
или
дефектны молеку-
I'llh-IIIa,
являющиеся рецептором
для
фибриногена
и
поэтому необходи-
1лн агрегации
со
всеми естественными агрегирующими агентами.
Следует
1
ить
внимание
на то, что при
изменениях этого рецептора нарушена также
И
ищрм'шая агрегация, поскольку
и для нее
необходимо образование фибрино-

геновых соединений
между
названными гликопротеиновыми рецепторами мри
лежащих тромбоцитов.
Нарушение механизма обратимой агрегации может быть связано также с <н
сутствием
плазменного компонента агрегации — фибриногена, а не мест фикщ
ции
его на плазматической тромбоцитарной мембране. Это приводит к схо.
с тромбастенией картине агрегационных нарушений, причина которых легко
выявляется на основании коагуляционных исследований, в
результате
которые
устанавливается афибриногенемия.
Для обеих форм патологии в фазе обратимой агрегации, связанных как •
собственно тромбоцитарным дефектом при тромбастении, так и с
отсутствием
плазменного кофактора агрегации при афибриногенемии, характерно выражен
ное
снижение ретракции фибринового
сгустка.
Это объясняется тем, что в гемо*
статической пробке нарушено формирование непрерывных тромбоцитар-
но-фибриногеновых/фибриновых
структур,
в которых сокращение актомиозинв
кровяных
пластинок приводит к сокращению системы только в
случае
сохранно-
сти
всех
ее компонентов.
Понятно,
что при афибриногенемии все отклонения в
тромбоцитарных
тестах
корригируются in
vitro
добавлением в систему фибрино-
гена, a in
vivo
— после повышения содержания фибриногена в крови до
0,5—1,0
г/л, 1
Выявление третьей основной группы патологии тромбоцитарного гемостаза,
в
основе которого лежит недостаточность секреторного процесса и вторичной
агрегации, проводится обычно на основании характерного резкого снижения
или
отсутствия второй волны агрегации с оптимальными дозами
всех
агреги-
рующих
агентов и единственной волны с коллагеном. Эти феномены, устанавли-
ваемые при графической регистрации агрегации тромбоцитов в приборах, явля-
ются
результатом
секреции из
гранул
хранения (плотных телец) этих клеток эн-
догенных
агрегирующих
агентов (АДФ, серотонина, адреналина,
Са
2
*).
В отличие от предыдущей группы нарушений, при тромбоцитопатиях высвобо-
ждения первая волна агрегации, вызываемая экзогенными агрегирующими аген-
тами, не изменена, поскольку нет дефектов ни во взаимодействии растворимых
агонистов с тромбоцитарными рецепторами, ни в доступности рецепторных мест
в
GPIIb-IIIa
и его связи с фибриногеном плазмы.
В настоящее время выделяют большое количество подгрупп тромбоцитопа-
тий
высвобождения, которые классифицируют по нарушению различных
меха-
низмов
активации тромбоцитов и реализации секреторного процесса. В каждой
из
них описано относительно небольшое число больных с врожденной пато-
логией, обычно с аутосомно-рецессивным типом наследования. В отличие от
вышеописанных
двух
групп нарушений тромбоцитарного гемостаза, тромбоци-
топатии высвобождения нередко развиваются в
результате
приобретенных де-
фектов
при различных основных заболеваниях и инфекционно-токсических
воздействиях на кровяные пластинки.
В обычных клинических условиях дифференцировать две основные подгруп-
пы
тромбоцитопатий высвобождения, обусловленных или дефектами в
путях
передачи сигналов активации, или дефицитом
гранул
хранения кровяных пла-
стинок,
в известной степени можно на основании электронно-микроскопиче-
ских исследований, а также оценки агрегации с арахидоновой кислотой и боль-
шими
дозами тромбина. Дефицит/дефект
гранул
хранения может быть
вызван или нарушением их наполнения в процессе кроветворения, или выра-
женной
активацией кровяных пластинок в циркуляции с последующим опусто-

i
хранении. Последнее наблюдается
в
патологических условиях,
пич
к
распитию тромбинемии
или
появлению
в
кровотоке высоких
кон-
iii иктнвирующих тромбоциты субстанций. Агрегация
с арахидоновой
• при
которой активация клеток опосредуется простагландин-тромбо-
i
путем после проникновения этого агента через тромбоцитарную плаз-
ую мембрану,
не
изменена
при
дефиците/дефекте гранул хранения
аимнаемом дефиците пула хранения),
так как
вторая волна агрега-
ппуется здесь через образующийся
ТхА
2
(первая положительная обрат-
I,),
.1 не
агонистами, высвобождаемыми
из
гранул хранения (вторая
по-
йми
обратная связь). Напротив,
при
дефиците пула хранения наруше-
•
i
рсцин
и
вторичная агрегация
с тромбином,
поскольку сигнал, переданный
юрез полифосфоинозитидный путь,
не
может завершиться секреци-
MVCTI.IX
или
опустошенных гранул.
при
(функциональной тромбоцитопатии высвобождения, которая
не*
миает обусловлена нарушениями передачи сигнала активации именно
.шдин-тромбоксановым путем, агрегация
с арахидоновой
кислотой
сни-
"|. шш
отсутствует,
так как
метаболизм этого агента нарушен из-за недоста-
|. in
ферментов данного пути.
В
этой ситуации, напротив, агрегация
с
силь-
•мистом тромбином,
как
правило,
не
изменена, поскольку
его
большие
при
нормальном наполнении гранул хранения эффективно активируют
• i но
полифосфоинозитидным путем. Исследование гранулярного аппарата
i<••<•
х пластинок при электронной микроскопии их ультрасрезов также мо-
действовать разграничению рассматриваемых
двух
типов тромбоцитопа-
1.к нобождения, выявляя снижение количества или плотности гранул хране-
т при дефиците пула хранения. Более точное определение вариантных форм
п.шой
группы тромбоцитопатии
требует
использования сложных биохи-
•
к
их методов.
Почти
при
всех
вышеописанных формах тромбоцитопатии вторично наруша-
ли и коагуляционная активность кровяных пластинок. Это объясняется тем, что
цмГчшшая перестройка, приводящая к стимуляции коагуляционной активности
•пцитов,
индуцируется как в период адгезивно-агрегационных реакций, так и
•мод секреции. Как правило, коагуляционная недостаточность является вто-
rtiM и связана с изменениями в
других
фазах тромбоцитарных превращений,
i
"испивая значимость описанных этапов лабораторного обследования боль-
ных с тромбоцитопатиями,
следует
отметить, что здесь проявляется известная
к
шюмерность относительно меньшей чувствительности общих скрининговых
т. которые нередко характеризуют эффект совокупности нескольких реак-
ции,
и связи с чем действие механизмов компенсации в них более вероятно. По-
гтму
чем более точно и избирательно
тест
связан с имеющимся у больного
и
||н'ктом, тем с большей вероятностью он выявляет его при слабой степени из-
мгмспия.
Нужно согласиться с высказанным R. M.
Biggs
и R. G. MacFarlane поло-
жением о том, что геморрагические проявления системного характера являются
мГи.ективным показателем нарушений гемостаза, и поэтому больные с кровото-
чивостью нуждаются в углубленном обследовании,
даже
при нормальных ре-
зультатах
первичных скрининговых тестов. Необходимо в разные периоды за-
'
и
шгиания, особенно при ухудшении состояния пациента, проводить повторные
MI
i недования с расширением их объема, с применением более адекватных и чув-
i
тигельных методов и, наконец, с обследованием кровных родственников.