Klipp E., Herwig R., Kowald A., Wierling C., Lehrach H. Systems Biology in Practice: Concepts, Implementation and Application
Подождите немного. Документ загружается.

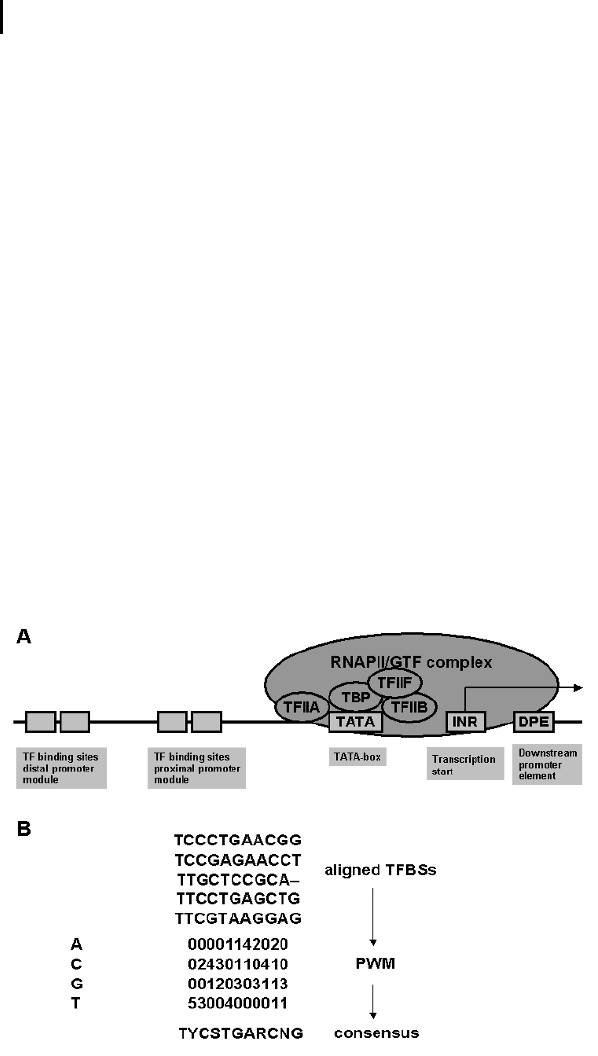
ditional proteins in order to transcribe the gene in the proper cellular context. In eu-
karyotes, gene expression requires a complex regulatory region that defines the tran-
scription start point and controls the initiation of transcription, i.e., the promoter.
Several algorithms are available that try to identify promoters for specific genes.
Some of these algorithms are discussed in this section.
8.2.1
General Promoter Structure
Promoter prediction algorithms implicitly assume a specific model for a typical pro-
moter. The general structure of an RNAP II promoter is described in Fig. 8.1a. The
typical promoter is composed of three levels of regulatory sequence signals. The first
level contains sequence motifs that enable the binding of specific transcription fac-
tors. The next level is the combination of binding sites to promoter modules that
jointly act as functional units. The third level consists of the complete promoter that
modulates gene transcription depending on cell type, tissue type, developmental
stage, or activation by signaling pathways.
The promoter must contain binding sites for the GTFs, such as the TATA box.These
proximate regulatory motifs constitute the core promoter that is able to bind the pre-
initiation complex and to determine the exact transcription start site. The core promo-
ter needs additional regulatory motifs at varying distances from the transcriptional
start point, the regulatory binding sites (transcription factor–binding sites, TFBSs).
These sites can be situated nearby or kilobases away from the core promoter.
260
8 Modeling of Gene Expression
Fig. 8.1 (a) General structure of a eukaryotic gene promoter. (b) Ex-
ample of a positional weight matrix and a consensus sequence de-
rived from different transcription factor–binding sites.

Transcription initiation can be viewed as a process involving successive formation
of protein complexes. In the first step, transcription factors bind to upstream promo-
ter and enhancer sequence motifs and form a multiprotein complex. In the next
step, this complex recruits the RNAP II/GTF complex to the core promoter and the
transcription start site. This is done through protein-protein interactions either di-
rectly or by adaptor proteins (Ptashne and Gann 1997). The full complex then starts
the transcription process.
The core promoter is located in the direct neighborhood of the transcription start
site (approximately 30 bp). The core promoter is the best-characterized part of the
promoter and is defined as a set of binding sites sufficient for the assembly of the
RNAP II/GTF complex and for specifying transcriptional initiation. Several types of
core promoters are known (Berg and von Hippel 1987):
1. TATA box: If TBP is present in the RNAP II/GTF complex, then this protein binds
to the sequence motif and the transcription starts approximately 30 bp down-
stream.
2. TATA-less: No TATA box is present. The start site is determined by a sequence mo-
tif INR (initiator region) surrounding the start site (Smale 1994).
3. A combination of both INR and TATA box
4. Null promoter: Neither of the two sequence motifs is present. Transcription initia-
tion is based solely on upstream (or downstream) promoter elements (Novina and
Roy 1997).
5. In some cases, a downstream promoter element (DPE) exists in addition to INR,
and both elements are able to specify the transcription start site (Burke and Ka-
donga 1997).
Whereas the core promoter determines the transcription start site, this function
cannot explain how genes whose protein products are needed in parallel are co-regu-
lated, e.g., from genes that are located on different chromosomes. Thus, additional
regulatory elements are necessary that meet the requirement of higher flexibility
and coordinated gene expression.
Typically a few hundred base pairs upstream of the core promoter is the proximate
promoter module, which contains TFBSs for proteins responsible for the modula-
tion of the transcription. The corresponding factors can influence the binding of the
core promoter components or the chromatin structure (or both). Furthermore, a pro-
moter can contain a distal promoter module (on the order of kilobases apart from
the transcription start site). Although these modules cannot act as promoters on
their own, they are able to enhance or suppress the activity of transcription up to or-
ders of magnitude (enhancer or silencer). Enhancer and silencer often exhibit a tis-
sue-specific activity. Like the transcription factors binding to the proximate module
of the promoter, the factors binding to the distal module influence gene expression
by interactions with the factors in the RNAP II/GTF complex or by changing the
chromatin structure. There is no clear boundary for the promoter in the 5' direction,
and the common explanation for interactions with distal factors to the transcription
apparatus is given by the formation of large loops in the DNA. The function of a pro-
261
8.2 Promoter Identification

moter is to increase or repress the transcription from the core promoter (basal tran-
scription). Thus, any given gene will have a specific regulatory region determined by
the binding sites of the transcription factors that ensure that the gene is transcribed
in the appropriate cell type and at the proper point in development. The transcrip-
tional activation is determined not only by the presence of the binding sites but also
through the availability of the corresponding transcription factors. These transcrip-
tion factors are themselves subjected to regulation and activation, e. g., through sig-
naling pathways, and the whole process can entail complex procedures such as tran-
scriptional cascades and feedback control loops (Pedersen et al. 1999).
8.2.2
Sequence-based Prediction of Promoter Elements
This section discusses promoter prediction algorithms that incorporate solely the
genome sequence. As described in the previous section, promoters are complex and
diverse, which makes promoter prediction a difficult task. Early reviews on promoter
recognition programs can be found in Fickett and Hatzigeorgiou (1997) and Stormo
(2000); a more recent review on algorithms for promoter prediction can be found in
Werner (2003).
The modeling of gene transcription regulation follows its combinatorial nature,
starting from the detection of individual binding sites (5–25 bp in length), moving to
the detection of specific combinations of binding sites, so-called composite regula-
tory elements (Kel et al. 1995), and finally to the detection of the promoter.
The detection of individual binding sites is the first level in that process. TFBSs
have high sequence variability, which distinguishes them, for example, from restric-
tion sites, i.e., the recognition sequences of a restriction enzyme. Whereas restric-
tion sites are almost exact in the sense that sites varying by only a single mismatch
will be cut less well by orders of magnitude, transcription factor binding can tolerate
high sequence variability of the TFBSs (Stormo 2000). This variation makes biologi-
cal sense in that it allows a higher flexibility of the regulatory system and assigns the
promoters different activity levels.
In order to meet this flexibility, known TFBSs for the same transcription factor
that may vary slightly are often represented by a consensus sequence that is close to
each single motif according to some criterion. There is a tradeoff in the consensus
sequences between the number of mismatches that are allowed and the precision of
the representation and thus a tradeoff between the specificity and the sensitivity of
the algorithms. A consensus sequence is typically denoted in the IUPAC code to de-
scribe ambiguities in nucleotide composition (Fig. 8.1b).
An alternative to consensus sequences is the use of positional weight matrices
(PWM). A PWM is a matrix representation of a TFBS, with rows representing one of
the bases, “A,” “C,” “G,” and “T,” and columns representing the position within the
motif (Fig. 8.1 b). Each entry in the matrix corresponds to a numerical value indicat-
ing the confidence for the specific base at that position. The PWM approach is some-
what more general than the consensus sequence approach in the sense that each
consensus can be represented by a PWM (for example, through frequency counts
262
8 Modeling of Gene Expression

across the aligned motifs) such that the same set of sites can be matched but not
vice versa (Stormo 2000). The calculation of the matrix elements can be performed
in different ways. Stormo et al. (1982) applied a neural network learning algorithm
to determine the weights of a PWM to distinguish known sites from non-sites in a
training sample of E. coli sequences. Afterwards they predicted new sequences using
the calculated weights. Berg and von Hippel (1987) used thermodynamic considera-
tions to compute the weights of a PWM. They showed that the logarithms of the
base frequencies should be proportional to the binding energy contribution of the
bases, assuming an equal distribution of base pairs through the genome. The most
comprehensive collection of PWMs can be found in the TRANSFAC database.
Recognition of composite regulatory elements has been proposed in order to meet
the combinatorial nature of gene regulation, e.g., of two transcription factors that in-
teract with each other in gene regulation. Here, statistical approaches have been
made to reveal common pairs from DNA sequences. For example, Kel et al. (1999)
developed a method that employs pairs of weight matrices for two corresponding
transcription factors. The method takes into account the matching distances of the
matrices on the DNA sequence and the mutual orientation and combines this with
binding energy considerations. A number of examples of composite regulatory ele-
ments have been collected in the TRANSCompel database (Kel-Margoulis et al. 2002).
The general principle of promoter recognition methods is based on the strategy to
determine a promoter model by features that are trained on a set of known promoter
and non-promoter sequences. These features are subsequently used to search for an
unknown number of promoters in a contiguous DNA sequence. The methods are
distinguished from each other by the way the features are determined. Typically,
they fall into two groups. The first group uses the pure sequence composition and is
based on scoring moving sequence windows, whereas the second group employs
prediction based on the detection of motifs from the core promoter element such as
TATA box or INR.
The first group of algorithms can be exemplified by the PromFind method de-
scribed in Hutchinson (1996). This method is based on the idea of discriminative
counts of sequence groups. PromFind uses the frequency of heptamers in coding
and non-coding sequences trained on sequences of 300 bp in length. Discrimination
is based on the following measure:
d
i
s
f s
f sf
i
s
; i 1; 2 : (8-1)
Here, f (s) denotes the frequency of heptamer s in the promoter sequences and
f
i
(s) corresponds to the frequency of the heptamer in the training sample (i=1:
non-coding, i=2: coding). For each sequence in a window of size 300 bp, the two
measures are calculated and the window with the best score is returned. Another
way of computing discriminative counts is employed in PromoterInspector devel-
oped by Scherf et al. (2000).
The second group of algorithms uses biological sequence features from the core
promoter. Prestridge (1995) combined several of those patterns. The hit ratio of
263
8.2 Promoter Identification

known TFBSs within promoters and non-promoters is used as an indicator for the
identification of a promoter. The combined ratio scores of all TFBSs in a certain se-
quence window are used to build a scoring profile. This profile combined with a
weight matrix for TATA boxes is used for predicting the transcription start site. Other
methods model the core promoter with artificial neural networks (Reese 2001), en-
sembles of multi-layer perceptrons for binding sites or Hidden Markov models.
A list of some promoter recognition programs is found in the following table:
Program Web location Reference
FunSiteP http://compel.bionet.nsc.ru/FunSite/fsp.html Kondrakhin et al. (1995)
PomoterInspector http://www.genomatix.de/cgi-bin/ Scherf et al. (2000)
promoterinspector/promoterinspector.pl
PromoterScan http://bimas.dcrt.nih.gov/molbio/proscan Prestridge (1995)
NNNP http://www.fruitfly.org/seq_tools/promoter.html Reese (2001)
PromFind http://iubio.bio.indiana.edu/soft/molbio/mswin/ Hutchinson (1996)
mswin-or-dos/profin11.exe
TSSG/TSSW http://www.softberry.com Solovyev and Salamov
(1997)
FirstEF http://rulai.cshl.org/tools/FirstEF Davuluri et al. (2001)
8.2.3
Approaches that Incorporate Additional Information
Since it has been shown that the error rates of the promoter prediction programs are
fairly unsatisfactory (Fickett and Hatzigeorgiou 1997), new developments are trying
to incorporate additional information as a backup when predicting TFBSs. A first
class of approaches combines binding site prediction with gene expression data de-
rived from DNA arrays. The widespread use of DNA arrays (cf. Chapter 9) has given
rise to the following general program: (1) identify co-expression groups by clustering
or other statistical methods and (2) search in the upstream regions of the grouped
genes for common regulatory motifs. This approach was utilized for the first time by
Tavazoie et al. (1999) for identifying novel regulatory networks in Saccharomyces cere-
visiae. The authors used a K-means clustering algorithm to identify groups of co-
regulated genes. They identified common sequence motifs in the upstream se-
quences of the genes and identified 18 motifs in 12 clusters that were highly over-re-
presented within their own cluster and absent in the others, thus indicating the exis-
tence of different regulation patterns.
This and other studies (Pilpel et al. 2001) have demonstrated that genes that are
co-expressed across multiple experimental conditions underlie common regulatory
mechanisms and thus share common TFBSs in their promoters. Although these re-
sults are promising, methods that work well in yeast are difficult to extend to higher
eukaryotes. This is mainly due to the fact that in yeast regulatory sequences are fairly
proximal to the transcription start site, whereas in higher eukaryotes these se-
quences can be located many kilobases on either side of the coding region. A recent
264
8 Modeling of Gene Expression

approach to human data has been published (Elkon et al. 2003). Here, the authors
used DNA array data and human genome sequence data to identify putative regula-
tory elements that control the transcriptional program of the human cell cycle. They
identified several transcription factors (such as E2F, NF-Y, and CREB) whose regula-
tory sequences were enriched in cell cycle–regulated genes and assigned these fac-
tors to certain phases of the cell cycle.
A second class of approaches uses comparative sequence analysis from upstream
sequences of orthologous genes through different organisms (Wassermann et al.
2000). These authors investigated skeletal muscle–specific transcription factors and
found that their binding sites are highly conserved in human and mouse DNA se-
quences. The general observation of conserved non-coding regions throughout dif-
ferent organisms has given rise to a number of recent developments that incorporate
cross-species analysis of promoter elements. For example, Dieterich et al. (2003)
have developed a comparative approach to human and mouse regulatory regions
and built up a database of so-called conserved non-coding sequences (CORG, http://
corg.molgen.mpg.de/).
A combination of these two approaches has been applied to the detection and ex-
perimental verification of a novel cis-regulatory element involved in the heat shock
response in C. elegans (Thakurta et al. 2002). The authors identified co-regulated
genes with DNA arrays and investigated the upstream regions of these genes for pu-
tative binding sites by pattern recognition algorithms. In the case of either signifi-
cant over-representation or cross-species conservation, they build biological assays of
the regulatory motifs using GFP reporter transgenes.
Additional sequence information is also sometimes incorporated in promoter
identification, in particular the identification of CpG islands. It has been reported
that these CpG islands correlate with promoters in vertebrates so that their features
are used in the computational process. By definition (Gardiner-Garden and From-
mer 1987) CpG islands are genomic regions that (1) are longer than 200 bp, (2) have
nucleotide frequencies of C and G in that region greater than 50%, and (3) have
CpG dinucleotide frequency in that region higher than 0.6 of that expected from
mononucleotide frequencies.
Despite all these developments, the recognition and identification of promoter ele-
ments remain error prone due to the highly complex nature of eukaryotic gene regu-
lation. Future approaches thus will have to incorporate additional information to a
much larger extent than is currently done.
8.3
Modeling Specific Processes in Eukaryotic Gene Expression
We want to know which genes are expressed, to what level, and where and when in
order to comprehend the functioning of organisms at the molecular level. A network
of interactions among DNA, RNA, proteins, and other molecules realizes the regula-
tion of gene expression. This network involves many components. There is forward
flow of information from gene to mRNA to protein according to the dogma of mole-
265
8.3 Modeling Specific Processes in Eukaryotic Gene Expression

cular biology. Moreover, positive and negative feedback loops and information ex-
change with signaling pathways and energy metabolism ensure the appropriate reg-
ulation of the expression according to the actual state of the cell and its environment.
Modeling of gene expression is an example of a scientific field where one may ob-
tain results with different techniques. The dynamics or the results of gene expres-
sion have been mathematically described with Boolean networks, Bayesian net-
works, directed graphs, ordinary and partial differential equation systems, stochastic
equations, and rule-based formalisms.
Although understanding of the regulation of large groups of genes, of the emer-
gence of complex patterns of gene expression, and of relations with inter- and intra-
cellular communication is still a scientific challenge, many insights have already
been gained from the modeling of particular processes or of the regulation of indivi-
dual sets of genes.
8.3.1
One Example, Different Approaches
In the following sections we will present an overview of modeling approaches and
the scientific questions that can be tackled with different techniques. For the sake of
clarity, we will use only examples with a low number of components (genes and pro-
teins), although the presented approaches can also be applied to larger systems.
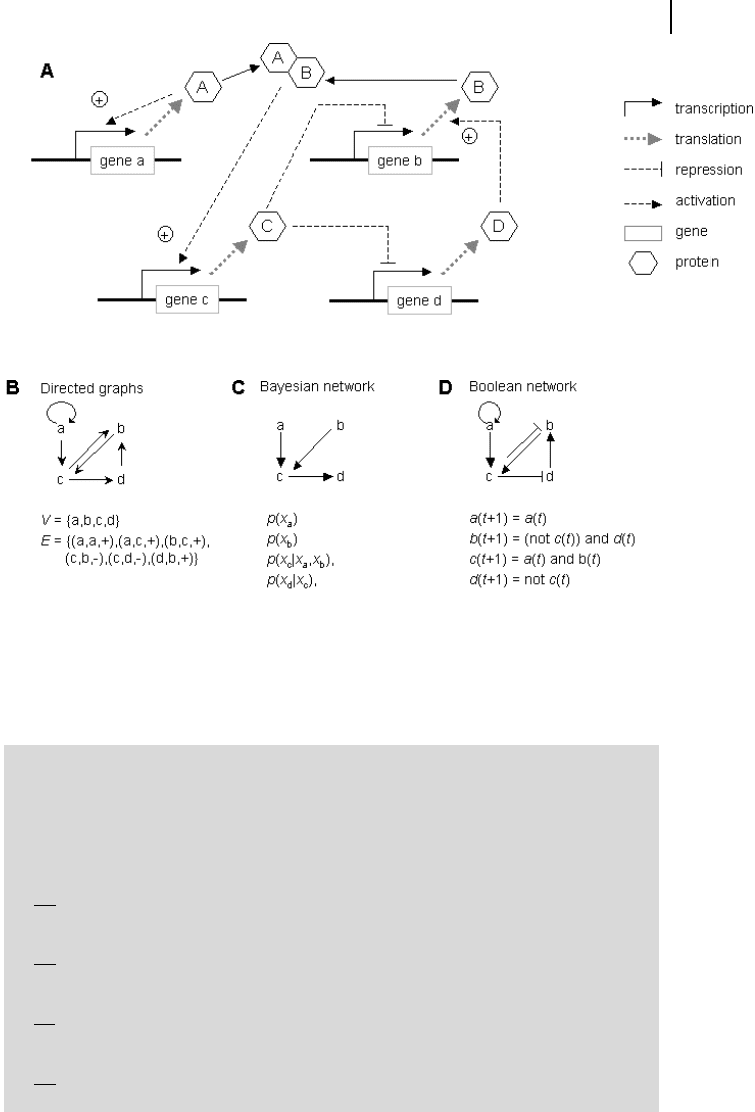
The example presented in Fig. 8.2 contains four genes, a through d, which code
for the proteins A through D. mRNA is not shown for sake of simplicity. The pro-
teins A and B may form a heterodimer that activates the expression of gene c. Pro-
tein C inhibits the expression of genes b and d, which are in this way co-regulated.
Protein D is necessary for the transcription of protein B.
8.3.1.1 Description with Ordinary Differential Equations
Gene expression can be mathematically described with systems of ordinary differen-
tial equations in the same way as dynamical systems in metabolism (Chapter 5), sig-
naling (Chapter 6), and other cellular processes (Chapter 7). In general, one considers
dx
i
dt
f
i
x
1
; ::; x
n
i 1; :::; n : (8-2)
The variables x
i
represent the concentrations of mRNAs, proteins, or other mole-
cules. The functions f
i
comprise the rate equations that express the changes of x
i
due
to transcription, translation, or other individual processes. For details about how to
specify the rate equations and how to analyze the resulting ODE systems, compare
Sections 5.1, 5.2 and 3.2.
266
8 Modeling of Gene Expression
Example 8-1
The dynamics of the system depicted in Fig. 8.2 can be described in several ways
depending on the desired particularization. If we consider only the mRNA abun-
dances a, b, c, and d, we get:
da
dt
f
a
a
db
dt
f
b
b; c; d
dc
dt
f
c
a; b; c
dd
dt
f
d
c; d: (8-3)
267
8.3 Modeling Specific Processes in Eukaryotic Gene Expression
Fig. 8.2 Gene regulatory network comprising
four genes a–d. (a) Dependence of translation
of genes a–d, the transcription of their mRNAs
(not shown), and the influence of the respective
proteins A–D. (b) Representation as directed
graph. (c) Respective Bayesian network. Note
that some interactions are neglected (inhibition
of b by c, activation of b by d) in order to get a
network without cycles. (d) The Boolean net-
work.

Specific expressions for the functions f, which consider the depicted regulatory in-
teractions, could be the following:
f
a
av
a
k
a
a
f
b
b; c; d
V
b
d
n
d
K
b
d
n
d
K
Ic
c
n
c
k
b
b
f
c
a; b; c
V
c
a b
n
ab
K
c
a b
n
ab
k
c
c
f
d
c; d
V
d
K
Ic
c
n
c
k
d
d : (8-4)
Here, k
a
, k
b
, k
c
, and k
d
are the first-order rate constants of the degradation of a, b,
c, and d, respectively. v
A
denotes the constant rate of expression of gene a, and the
Hill term
V
b
d
n
d
K
b
d
n
d
describes the formation of b activated by d with maximal rate
V
b
, dissociation constant K
b
, and Hill coefficient n
d
. The inhibition by c is ex-
pressed by the term (K
Ic
+ c
n
c
). The formation of c is modeled with a Hill expres-
sion that points to a threshold of the formation of c depending on the concentra-
tions of a and b. V
c
and K
c
are maximal rate and dissociation constant, respec-
tively, and n
ab
is the Hill coefficient. The production of d depends on the maximal
rate V
d
and on the inhibition by c. The dynamics for a certain choice of para-
meters is shown in Fig. 8.3.
The ODE formalism allows involving more details, e. g., the explicit consideration
of the protein concentrations. Considering specifically the mRNA of gene b and
protein B, we get
d
dt
b
V
b
K
Ic
C
n
c
k
b
b
d
dt
B D
V
B
b
K
B
b
k
B
B k
AB
A B : (8-5)
This means that we can distinguish between the processes determining the velo-
city of translation (basic rate V
b
and inhibition by protein C), transcription (depen-
dence on mRNA concentration b and on the activator concentration D), and de-
gradation or consumption on both levels (degradation of b and B and formation
of complex AB).
The advantage of the description with ODE systems is that one can take into ac-
count detailed knowledge about gene regulatory mechanisms such as individual ki-
netics, individual interactions of proteins with proteins or proteins with mRNA, and
so on. A profound disadvantage is the current lack of exactly this type of knowledge
– the lack of kinetic constants due to measurement difficulties and uncertainties in
the function of many proteins and their interactions.
268
8 Modeling of Gene Expression
The ODE formalism allows consideration of many specific aspects of gene regula-
tion and cellular physiology. Time delay in gene regulation due to the involvement of
many different and comparatively slow processes can be considered either by using
delay differential equations or by considering all slow processes individually (see
Section 8.3.2). Dilution of compounds due to cell growth is usually considered by
adding a dilution term.
8.3.1.2 Representation of Gene Network as Directed and Undirected Graphs
A directed graph G is a tuple [V, E], where V denotes a set of vertices and E a set of
edges (cf. Section 3.5). The vertices i BV correspond to the genes (or other compo-
nents of the system) and the edges correspond to their regulatory interactions. An
edge is a tuple [i, j] of vertices. It is directed if i and j can be assigned to the head
and tail of the edge, respectively. The labels of edges or vertices may be expanded to
contain information about the genes and their interactions. In a general way, one
may express an edge as a tuple [i, j, properties]. The entry properties can simply indi-
cate whether j activates (+) or inhibits (–) i (Fig. 8.2 b). The entry properties can also
be a list of regulators and their influence on that specific edge, such as [i, j,((k, acti-
vation), (l, inhibition as homodimeric protein))].
In principle, many databases that provide information about genetic regulation
are organized as richly annotated directed graphs (e. g., Transfac, KEGG; see Chapter
13). Directed graphs are not suited to predict the dynamics of a network, but they
may contain information that allows certain predictions about network properties:
. Tracing paths between genes yields the sequence of regulatory events, shows re-
dundancy in the regulation, or indicates missing regulatory interactions (that are,
for example, known from experiment).
. A cycle in the network may indicate feedback regulation.
. Comparison of gene regulatory networks of different organisms may reveal evolu-
tionary relations and reveal targets for bioengineering and for pharmaceutical ap-
plications (Dandekar et al. 1999).
. The network complexity can be measured by the connectivity, i.e., the distribution
and the average of the numbers of regulators per gene.
269
8.3 Modeling Specific Processes in Eukaryotic Gene Expression
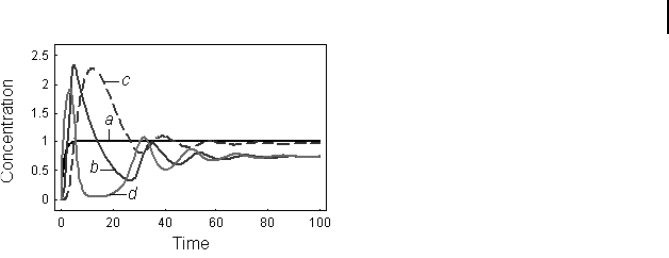
Fig. 8.3 Dynamics of the mRNA concentra-
tions of the system presented in Example 8-1
according to Eq. (8-4). Parameters: v
a
=1,
k
a
=1,V
b
=1,K
b
=5,K
Ic
= 0.5, n
c
=4,k
b
= 0.1,
V
c
=1,K
c
=5,k
c
= 0.1,V
d
=1,k
d
= 1. Initial
conditions: a(0) = b(0) = c(0) = d(0) = 0.